#11. Médulloblastomes de l’adulte
Jacques GUYOTAT, M.D., Thiébaud PICART, M.D., CHU Lyon, mars 2022.
Questions
1. Concernant l’épidémiologie et la présentation clinique des médulloblastomes de l’adulte, quelle(s) proposition(s) est/sont exactes ?
A. Le médulloblastome fait partie des tumeurs cérébelleuses les plus fréquemment diagnostiquées chez l’adulte.
B. Les médulloblastomes de l’adulte sont rarement révélés par un syndrome cérébelleux .
C. En post opératoire une PL doit être systématiquement réalisée avant une IRM spinale
D. Les formes pédiatriques et les formes adultes sont très différentes d’un point de vue clinique, moléculaire, génétique et pronostique.
E. Les médulloblastomes de localisation vermienne peuvent être révélés par une atteinte des nerfs mixtes.
2. Concernant la classification histo-moléculaire des médulloblastomes, quelle(s) proposition(s) est/sont exactes ?
A. Les médulloblastomes sont divisés en 4 groupes histologiques et 4 groupes moléculaires qui sont strictement concordants.
B. Les médulloblastomes desmoplasiques/nodulaires présentent habituellement une activation de la voie WNT.
C. Les médulloblastomes du groupe moléculaire 3 sont de bon pronostic.
D. Les médulloblastomes anaplasiques/à grandes cellules sont parfois associés au syndrome de Gorlin.
E. Des altérations du chromosome 17 sont fréquemment retrouvées dans les médulloblastomes du groupe 4.
3. Parmi les caractéristiques radiologiques suivantes, laquelle/lesquelles est/sont compatibles avec le diagnostic de médulloblastome chez un patient présentant une tumeur cérébelleuse ?
A. Hydrocéphalie tétraventriculaire
B. Prise de contraste hétérogène
C. Restriction de diffusion
D. Augmentation majeure du rCBV
E. Nodules disséminés au niveau des racines de la queue de cheval
4. Parmi les médulloblastomes suivants, lequel/lesquels est/sont à haut risque ?
A. Médulloblastome vermien, classique, du groupe WNT, sans résidu tumoral, avec la présence de cellules tumorales dans le LCS mais sans nodule suspect à distance de la tumeur
B. Médulloblastome vermien, classique, du groupe WNT, avec un résidu tumoral de 3 cm3, adhérent au plancher du V4 et sans métastase
C. Médulloblastome hémisphérique droit, desmoplasique, SHH-activé et TP53 muté, avec un résidu tumoral de 1cm3 et sans métastase
D. Médulloblastome hémisphérique gauche, desmoplasique, SHH-activé et TP53 sauvage, avec un résidu tumoral de 1cm3 et avec la présence d’un nodule de 5 mm prenant le contraste et localisé en regard de T12
E. Médulloblastome vermien, classique, du groupe 3, sans résidu tumoral post-opératoire, sans métastase
5. Concernant la prise en charge des médulloblastomes de l’adulte et le pronostic, quelle(s) proposition(s) est/sont exacte(s) ?
A. Les complications neurologiques post-opératoires peuvent consister en un syndrome cérébelleux, des troubles de la déglutition ou un mutisme akinétique
B. La chirurgie d’exérèse doit toujours viser à réaliser une exérèse complète voire supra-marginale
C. La réalisation d’une IRM post-opératoire est recommandée mais pas avant 48 heures afin d’évaluer la qualité de l’exérèse
D. Lorsque l’exérèse est complète, la réalisation systématique d’un traitement adjuvant n’est pas recommandée.
E. A l’issue du traitement, la surveillance clinique et radiologique doit être poursuivie pendant au moins 10 ans.
I. Epidémiologie
Le médulloblastome (MB) est une tumeur cérébrale maligne primitive localisée à la fosse cérébrale postérieure. Chez l’enfant (réf.4) cette lésion constitue entre 15% et 25% des tumeurs cérébrales primitives du système nerveux central (1). Chez l’adulte le MB est très rare puisqu’il ne représente que 1% des tumeurs intracrâniennes. L’incidence annuelle est estimée à 0,6 par million d’habitants avec une prédominance entre 20 et 40 ans et chez les sujets de sexe masculin (2,3).
La majorité des MB sont sporadiques mais certaines formes sont diagnostiquées dans le cadre de pathologies génétiques : syndrome de Turcot , de Li Fraumeni et Gorlin dans les MB SHH , polypose colique familiale dans les MB WNT (4). Les formes de l’enfant et de l’adulte sont considérées comme différentes sur le plan épidémiologique, clinique, moléculaire, génétique et pronostique (5–8). La rareté de MB de l’adulte explique que les protocoles de traitement sont souvent inspirés de ceux de l’enfant et que leur efficacité reste difficile à évaluer avec un niveau de preuve élevé (9,10).
II. Classification histo-moléculaire
Les MB sont des tumeurs neuroépithéliales embryonnaires de grade IV selon la classification de l’OMS 2021. La classification des MB identifie 4 groupes histopathologiques et 4 groupes moléculaires qui ne sont pas strictement concordants. L’intégration des données histologiques et moléculaires est nécessaire pour évaluer le pronostic à l’échelle individuelle (11–13).
1. Classification histologique
La classification histologique de l’OMS 2016 distingue 4 groupes (11,12) :
- MB classiques : 65% à 70% des cas
Il s’agit du type histologique le plus fréquent, pouvant être associé à chacun des groupes moléculaires. A l’examen microscopique, il est possible d’observer des rosettes neuroblastiques de Homer-Wright, une disposition palissadique des cellules et parfois des nodules clairs à différenciation neurocytique (MB classique avec nodules). Ces tumeurs sont préférentiellement médianes. - MB desmoplasiques/nodulaires : 20% des cas
Les MB desmoplasiques/nodulaires se caractérisent par la présence de zones nodulaires claires, séparées par un réseau de fibres collagéniques et dépourvues d’organisation en rosettes neuroblastiques. Ces tumeurs peuvent être médianes ou hémisphériques. Elles sont généralement associées à une activation de la voie SHH et surviennent parfois dans le cadre d’un syndrome de Gorlin (naevomatose basocellulaire) (14). - MB anaplasiques/à grandes cellules : 10% des cas
Ils se caractérisent par la présence de critères d’anaplasie. La présence de grandes cellules s’associe fréquemment aux critères d’anaplasie, ce qui a conduit à regrouper les MB anaplasiques et les MB à grandes cellules dans une même entité. Ils peuvent s’associer à tous les groupes moléculaires mais sont plus fréquents dans le groupe SSH-activé et dans le groupe 3. Ils sont souvent métastatiques d’emblée et associés à un mauvais pronostic (11,12). - MB à nodularité extensive : 3% à 4% des cas
Ils se caractérisent par la présence de nombreux nodules clairs, de grande taille, constitués par des cellules neurocytiques, peu proliférantes. Le contingent cellulaire internodulaire est plus réduit que dans les MB desmoplasiques. Ils sont très rares chez l’adulte et ont un excellent pronostic. Ils sont généralement associés à une activation de la voie SHH. A l’instar des formes desmoplasiques/nodulaires, ils peuvent être associés au syndrome de Gorlin. Ils sont de localisation essentiellement vermienne (11,13,15).
2. Classification moléculaire
La classification moléculaire distingue 4 entités :
- MB WNT-activés : 15% des cas
Les MB WNT-activés correspondent histologiquement à des MB classiques ou exceptionnellement à des MB anaplasiques/à grandes cellules. Ces tumeurs sont associées à un excellent pronostic à tous les âges, surtout chez l’enfant (bas risque) (11,16). - MB SHH-activé : 60% des cas
Les MB SHH-activés (voie sonic hedge hog) surviennent à tout âge. Ils sont divisés en deux sous-groupes définis par le statut du gène TP53 (12,17).
Les MB SHH-activés et TP53 mutés correspondent histologiquement à des MB classiques, anaplasiques/à grandes cellules ou très rarement à des MB desmoplasiques/nodulaires. Ces tumeurs sont rares, surviennent chez le grand enfant et se caractérisent par leur mauvais pronostic indépendamment de la présence ou non de métastases (haut risque) (7,17).
Les MB SHH-activés et TP53 sauvage peuvent correspondre à tous les sous-types histologiques décrits et sont prédominants. Diverses altérations génétiques mutuellement exclusives et activant la voie SHH sont décrites. D’autres anomalies peuvent être associées (mutation du promoteur de TERT, amplification de N-MYC, délétion du 9q, perte de 17p). Ces tumeurs surviennent à tout âge et peuvent être de bas risque (formes desmoplasique/nodulaire ou à nodularité extensive), de risque standard (forme classique) ou de haut risque (amplification de NMYC, présence de métastases) (7,8,17).
- MB groupe 3 : 5% des cas
Les MB du groupe 3 correspondent histologiquement à des MB classiques ou anaplasiques/à grandes cellules. Ils présentent plus fréquemment une amplification de MYC que les MB du groupe 4. Ces tumeurs concernent essentiellement le petit enfant (<4 ans) et l’enfant (4-16 ans) et sont très rares chez l’adulte. Elles ont un mauvais pronostic, en particulier chez le petit enfant, lorsqu’il s’agit d’une forme anaplasique/à grandes cellules ou en présence de métastases (très haut risque) (12,18,19).
- MB groupe 4 : 20% des cas
Les MB du groupe 4 correspondent histologiquement à des MB classiques ou anaplasiques/à grandes cellules. Ils présentent fréquemment des altérations du chromosome 17 et des altérations des gènes KDM6A et SNCAIP. Ces tumeurs sont de risque standard sauf en présence de métastases (haut risque) (7,8,19,20).
III. Clinique
Les symptômes révélateurs sont ceux de toute tumeur de la fosse cérébrale postérieure. Le délai entre les premiers signes et le diagnostic est en moyenne de 3 mois mais peut varier de quelques semaines à 18 mois (21). La symptomatologie initiale est volontiers frustre, dominée par des céphalées, des nausées, des vertiges, une ataxie, plus rarement une atteinte des nerfs crâniens ou des troubles cognitifs (22). Au moment du diagnostic une hypertension intracrânienne allant de la simple céphalée aux troubles de conscience, souvent secondaires à une hydrocéphalie, est constatée chez près de 80% des patients. Le syndrome cérébelleux est beaucoup plus présent que chez l’enfant, constaté dans 45 à 65% des cas. Il s’explique par la localisation cérébelleuse fréquente chez l’adulte. Une hypoacousie ou une atteinte des nerfs mixtes sont possibles dans les localisations très latérales (8,23,24). Un examen clinique complet à la recherche de signes évocateurs d’une diffusion lepto-méningée ou extra-névraxique (poumon, ganglions, os, foie, moelle osseuse) doit être systématiquement réalisé (25).
IV. Bilan paraclinique
1. Imagerie
a. Imagerie cérébrale
Elle précisera la localisation tumorale exacte et recherchera une extension dans le foramen de Luschka, possible dans 15% des cas, et au niveau du plancher du V4. Chez l’adulte les tumeurs sont décrites comme cérébelleuses dans 50% à 90% des cas (26–28). L’imagerie recherchera aussi l’existence d’une hydrocéphalie et la présence éventuelle de métastases intracrâniennes ; notamment au niveau du vermis, des citernes de la base, de la région sous épendymaire des ventricules latéraux et du plancher du V3 (27). Au diagnostic, la tumeur est relativement volumineuse, entre 3 et 6 cm, et exerce un effet de masse sur le V4, à l’origine d’une hydrocéphalie chez 50% à 95% des patients. L’œdème est habituellement modéré. Des zones kystiques ou nécrotiques sont notées dans 30% des cas, des calcifications dans 25% et un saignement dans 10% des cas (27).
Le scanner cérébral sans et avec injection est souvent le premier examen réalisé, notamment dans le cadre de l’urgence, pour diagnostiquer une hydrocéphalie qui pourrait justifier d’un geste de drainage. La tumeur est le plus souvent spontanément hyperdense, rehaussée par le contraste (29).
L’IRM multimodale reste bien évidemment l’examen de choix. En T1, le MB est en iso ou hyposignal dans 90% des cas. En T2, il apparait en hypersignal T2 dans 2/3 des cas et en iso ou en hyposignal dans 1/3 des cas (28). Après injection, la prise de contraste est intense de manière plus ou moins homogène chez la moitié des patients et faible ou absente chez l’autre moitié (28). Sur les séquences de diffusion, il existe le plus souvent une restriction de la diffusion, avec un abaissement du coefficient de diffusion apparente. L’IRM de perfusion retrouve une augmentation modérée du rCBV autour de 1, témoignant d’une faible néoangiogénèse (30). Enfin, sur la spectroscopie, les pics de choline et de taurine sont élevés et celui du N-acétyl aspartate diminué (27).
Une étude récente a comparé les différences enfant/adulte. La localisation cérébelleuse, la prise de contraste peu intense et inhomogène, l’absence d’hyposignal sur la séquence T1 sont statistiquement plus fréquents chez l’adulte (26). Des corrélations ont été établies entre l’IRM et les sous-groupes histo-moléculaires. Ainsi, les MB SHH mutés sont principalement localisés dans le cervelet, s’accompagnent de plus d’œdème et ont une forte restriction de diffusion. Les MB WNT sont plus médians mais une extension est possible à l’angle ponto cérébelleux et ils ont tendance à saigner (31,32). Le groupe 4 est localisé au V4, parfois kystique, prend peu le contraste sauf dans les formes de mauvais pronostic et dissémine volontiers (33).
b. IRM médullaire
Elle vise à rechercher une diffusion leptoméningée qui est présente dans 11% à 43% des cas. Elle doit être systématiquement réalisée, soit, et de préférence, avant l’exérèse chirurgicale si le diagnostic a été évoqué, soit 15 jours après le geste pour éviter des faux positifs liés à l’inflammation post-opératoire et à la présence de sang (5,21,27,34). Le diagnostic est évoqué devant des prises de contraste nodulaires le long de l’axe médullaire, du cône et des racines de la queue de cheval (5,27,34). La présence de métastases au diagnostic est moins fréquente que chez l’enfant (7% versus 30%) et les métastases au cours de l’évolution surviennent plus précocement chez l’enfant que chez l’adulte 20 mois versus 36 mois (21,35).
c. Un bilan paraclinique plus complet (scanner TAP, PET-scan, myelogramme…)
Il ne se justifie qu’en cas de signes cliniques évocateurs de métastases extra-névraxiques (poumon, ganglions os, foie, moelle osseuse), présentes dans 3% à 7% des cas (5,25,34).
2. Ponction lombaire
La recherche d’une dissémination lepto-méningée doit par ailleurs systématiquement comporter une ponction lombaire à réaliser après l’IRM spinale du fait de la possibilité de faux positifs post PL (5,36).
IV. Principe de prise en charge
La prise en charge des MB de l’adulte repose sur l’association chirurgie, radiothérapie et chimiothérapie à discuter en fonction de l’état clinique du patient et du stade risque standard versus haut grade. Les décisions doivent être prise de façon collégiale, en réunion de concertation pluridisciplinaire (RCP AJA) (5).
1. Traitement chirurgical
La chirurgie est la première étape de la prise en charge. L’objectif est d’obtenir un diagnostic histo-moléculaire précis et de réaliser une exérèse la plus complète possible tout en préservant l’état fonctionnel. Le drainage d’une hydrocéphalie, le plus souvent par ventriculocisternostomie, est le premier geste à réaliser, parfois en urgence en cas d’hydrocéphalie mal supportée. L’exérèse microchirurgicale sera précédée d’une analyse soigneuse des différentes séquences de l’IRM cérébrale pour préciser le volume, la localisation et tenter de prédire les difficultés auxquelles le chirurgien peut être confronté.
La position opératoire dépend avant tout des habitudes de l’équipe chirurgicale et anesthésique (37). La position assise a l’avantage d’être plus anatomique pour le chirurgien et de faciliter l’exérèse en diminuant la pression veineuse. Toutefois la majorité des centres l’ont abandonnée en raison des risques d’embolie gazeuse à l’origine de graves complications et de pneumatocèle systématique (38). Les positions en décubitus ventral ou en position dite « concorde », beaucoup plus rarement en décubitus dorsal avec rotation de la tête en cas de localisation très latérale, sont actuellement plus utilisées (39).
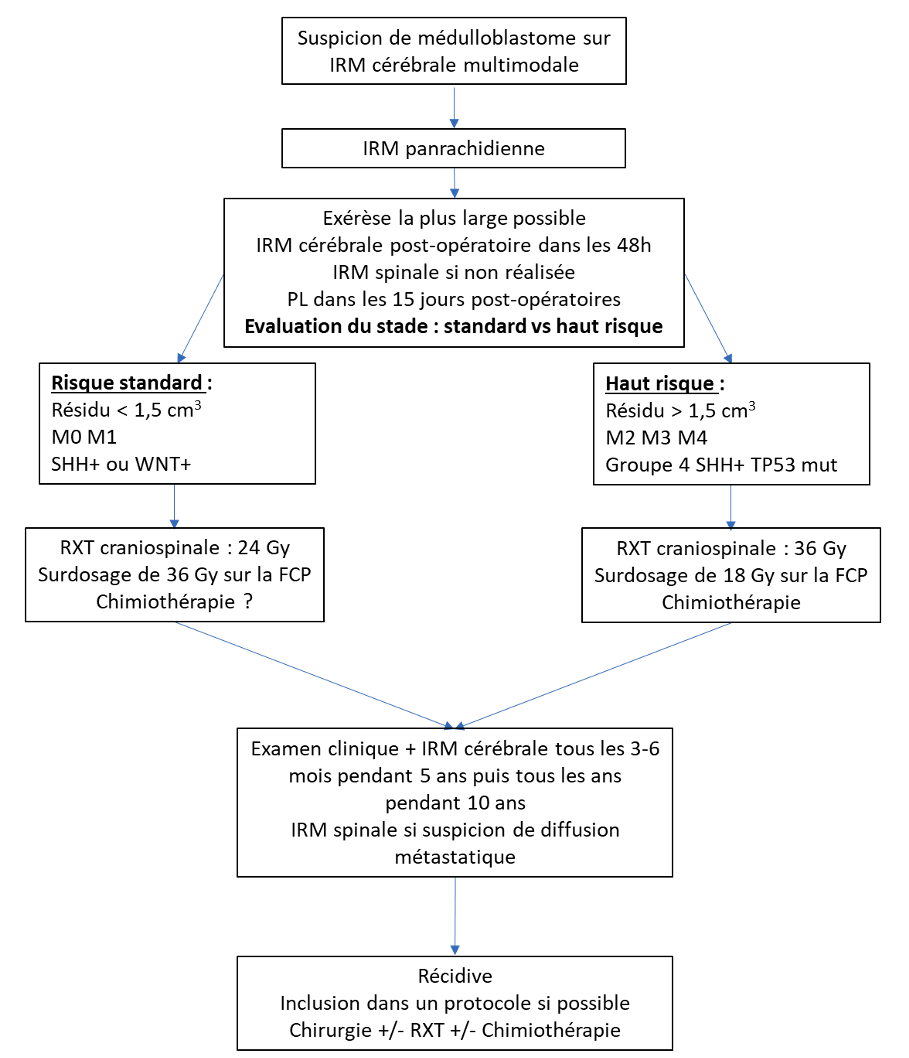
Après réalisation du volet, éventuellement complété d’une résection de C1 en cas de tumeur volumineuse à extension caudale, la grande citerne est ouverte. L’approche télo-velaire est recommandée (40). L’incision du vermis doit être limitée pour minimiser le risque de mutisme akinétique. Dans les localisations purement cérébelleuses, le cervelet sera incisé transversalement dans les sillons. Des rétracteurs sont souvent nécessaires pour exposer la tumeur. Celle-ci est habituellement molle et peu hémorragique, ce qui permet de l’aspirer et de l’évider à l’aspirateur ultrasonique. Le pôle supérieur est repéré puis le plancher du V4 qui doit représenter le plan à suivre sur la ligne médiane. Latéralement, la dissection se fait dans un plan sous-pial. Les difficultés sont principalement liées à une éventuelle infiltration du plancher du V4, des pédoncules cérébelleux et à une extension au foramen de Luschka qui sont les limites à une exérèse tumorale complète ou subtotale, estimée possible dans 60% à 85% des cas (5,21,41). Les complications sont l’ataxie cérébelleuse, la dysarthrie, l’atteinte des nerfs crâniens (troubles oculomoteurs et de déglutition). Le mutisme akinétique est beaucoup plus rarement décrit que chez l’enfant (42). Une IRM post opératoire doit être systématiquement réalisée dans les 48h afin de préciser la qualité de l’exérèse (résidu supérieur ou inférieur à 1,5 cm3), classer la tumeur en haut risque versus bas risque et préciser le type de traitement complémentaire (5).
2. Evaluation du stade
Les MB de l’adulte sont séparés en 2 groupes dont la valeur pronostique et la prise en charge sont différentes. Cette classification s’inspire de celles de Chang (43) et de Parker (44), reconnues chez l’enfant, plus récemment complétées par les données histo-moléculaires selon la classification OMS de 2016 (45).
MB à risque standard : résidu tumoral inférieur à 1,5 cm3, M0 (absence de métastase) ou M1 (présence de cellules dans le liquide cérébrospinal), de type classique ou desmoplastique et de sous-type moléculaire WNT ou SHH.
MB à haut risque : résidu supérieur à 1,5cm3, M2, M3, M4, sous-type moléculaire non WNT ou non SHH ou SHH TP53 muté.
3. Traitement complémentaire
La rareté de cette pathologie chez l’adulte explique l’absence de séries importantes qui, de plus, s’étalent sur plusieurs années et incluent différentes lignes de traitement. En pratique les protocoles de traitement chez l’adulte dérivent de ceux de l’enfant, d’analyses rétrospectives de cohortes mélangées enfants-adultes ou d’essais prospectifs non randomisés (10,25,26,46).
a. Radiothérapie
Le MB est une tumeur radiosensible. Une irradiation cranio-spinale par photons en modulation d’intensité (IRMT) ou par arc thérapie volumétrique modulée (VMAT) sera systématiquement réalisée par des équipes spécialisées. La dose préconisée est de 36 Gy en fractions quotidiennes de 1,8 Gy ou de 35,2 Gy en fractions quotidiennes de 1,6 Gy, 5 jours par semaine. Un surdosage sur la fosse cérébrale postérieure de 18 Gy à 20 Gy en 10 fractions pour un total de 54 ou 55,8 Gy sera associé (25,34). Chez l’adulte, la délivrance sur le lit tumoral seul n’impacterait pas le pronostic dans les MB à risque standard (9). Le traitement doit être débuté si possible dans les 4 à 6 semaines qui suivent la chirurgie et sans interruption (5,10,34,41). A l’instar de ce qui a été proposé chez l’enfant, une diminution de la dose jusqu’à 23,4 Gy sur l’axe cranio spinal, pour réduire la toxicité à long terme de la radiothérapie notamment sur le plan neurocognitif, associée à de la chimiothérapie, semble possible sans perte d’efficacité dans les MB M0 et M1 (9,25,41). Chez certains patients métastatiques à mauvais pronostic, une radiothérapie hyperfractionnée associée à une chimiothérapie peut être proposée (20,47). Dans la même optique, une irradiation cranio-spinale par protons, pour réduire la toxicité post-radique, est une alternative (48,49).
b. Chimiothérapie
Dans les MB à haut risque, la chimiothérapie sera systématiquement réalisée car elle impacte favorablement la survie (10,21,41). Dans les formes à risque standard, l’efficacité de la chimiothérapie est plus discutée (49,50). Toutefois, la majorité des études récentes suggère un bénéfice sur la survie sans récidive et globale de la chimiothérapie associée à la radiothérapie comparée à la radiothérapie seule (5,21,47,51–53). Le moment de la chimiothérapie néoadjuvante, concomitante ou adjuvante, le nombre de cycles et le type de molécule ne font pas l’objet de consensus rendant l’interprétation des résultats difficile (6,34,41). La chimiothérapie néoadjuvante s’est révélée inefficace dans une étude récente avec une survie sans récidive et globale inférieure à une radiothérapie seule et à une radiothérapie associée à une chimiothérapie (21). Le protocole le plus utilisé en Europe est dérivé de celui de Parker pour l’enfant et associe une radiothérapie concomitamment à de la VINCRISTINE suivie de 8 cycles de LOMUSTINE/VINCRISTINE/CISPLATINE ou CARBOPLATINE (44). La chimiothérapie concomitante n’est pas systématiquement effectuée et/ou doit être modifiée car elle expose à une toxicité non négligeable (26). Le nombre de cycles théoriquement de 8 est réduit à 4 chez 30% des patients et les doses nécessitent d’être diminuées (26). En France l’association CARBOPLATINE/ETOPOSIDE avec 2 cycles avant et 2 cycles après la radiothérapie est proposée dans le cadre d’un essai de phase 2 (NCT01857453 Clinical trials.gov).
V. Résultats et facteurs pronostiques
Le pronostic des MB de l’adulte, comparativement à d’autres tumeurs cérébrales malignes, est bon avec une survie à 5 ans de 70% avec un traitement par radio chimiothérapie (5). Un certain nombre de facteurs pronostiques ont été décrits, parfois différents de ceux retrouvés chez l’enfant.
1. Age et sexe
Un meilleur pronostic est rapporté chez les patients âgés de 18 à 20 ans, comparativement à ceux de 21 à 40 ans et de plus de 40 ans mais non confirmé par d’autres (21,54). Cette différence apparait surtout lorsque le suivi est de plus de 4 ans (2). La valeur pronostique du sexe n’est pas claire (21,53). Certaines séries rapportent une survie significativement inférieure chez les hommes avec un taux de mortalité supérieur de 26% à celui des femmes (5,55).
2. Qualité de l’exérèse
La qualité de l’exérèse, tout comme l’extension de l’infiltration tumorale (T1-T4), qui limite cette dernière, sont des critères différenciant les MB standards des MB à haut risque et le type de traitement complémentaire. Même si l’impact d’un résidu tumoral sur la survie est discuté, une exérèse complète (résidu inférieur à 1,5 cm3) reste un facteur de bon pronostic dans plusieurs études notamment dans les sous-groupes moléculaires de grade 4 (8,25,46,53,56,57). Bien évidemment, cet objectif ne doit pas se faire aux dépends d’éventuelles séquelles qui impacteraient la qualité de vie des patients.
3. Bas risque versus haut risque
Dans les formes standards, la survie globale varie de 82% à 100% à 5 ans et de 55% à 100% à 10 ans. La survie sans récidive de 65% à 80% à 5 ans. Dans les formes à haut risque, la survie globale varie de 62% à 73% et la survie sans récidive de 39% à 69% à 5 ans (5,6,10,20,41,49,55).
4. Sous-type histologique
Le rôle pronostique des différents sous-groupes histo-moléculaires, bien démontré chez l’enfant et intervenant dans la prise en charge thérapeutique est plus discuté chez l’adulte (8,54,58). Les groupes WNT, SHH TP53 sauvage et groupe 4 ont un taux de survie à 5 ans respectivement de 80%, 70% et 50% (58–60). Ces différentes classes moléculaires ont toutes un pronostic moins satisfaisant que chez l’enfant. Les groupes SHH TP53 muté sont de moins bon pronostic que les SHH TP53 sauvage. Le profil cytogénétique a également un impact pronostique. La perte du chromosome 10q et le gain du chromosome 17 sont de mauvais pronostic (8,59,60).
5. Dissémination leptoméningée
Le stade M (MO-M4) au diagnostic a un rôle pronostique discuté. La présence de métastases au moment du diagnostic est un facteur de mauvais pronostic sur la survie globale et sans récidive dans certaines séries mais pas dans d’autres (20,21,50,52,56,61,62). Toutefois, les patients métastatiques reçoivent une chimiothérapie ce qui n’a pas forcément été le cas chez les patients non métastasiques (21).
VI. Suivi
Le suivi repose sur un examen clinique complet associé à une IRM cérébrale et spinale. L’objectif est de dépister le plus précocement possible une récidive et des complications liées au traitement. En l’absence de recommandations validées, la majorité des équipes recommandent une IRM cérébrale précoce à 48 h avec injection et séquence de diffusion pour évaluer la qualité de l’exérèse et détecter une complication post-opératoire puis un contrôle cérébral tous les 3 à 6 mois pendant 5 ans puis tous les ans pendant 10 ans (5,34,41). Une IRM médullaire est systématiquement associée pendant 2 ans pour certains ou simplement proposée éventuellement associée à une étude du liquide cérébrospinal en présence de métastases initiales ou de suspicion de dissémination leptoméningée (5,10,34). L’examen clinique et les bilans biologiques rechercheront d’éventuelles complications précoces ou secondaires liées à la chimiothérapie et à la radiothérapie : toxicité hématologique notamment chez les personnes âgées, auditive (47%), visuelle (cataracte), cutanée (20% d’alopécie), rénale, endocrinienne (53%) notamment de la fonction gonadotrope, neurologique (polyneuropathie notamment avec la vincristine) (10,22,46,63). Des séquelles cognitives tardives liées à la radiothérapie sont rapportées jusqu’à 70% des cas et devront être particulièrement recherchées compte tenu de l’impact psychosocial (22,63).
VII. Récidive
Les récidives surviennent entre 2 et 4 ans après la chirurgie initiale mais peuvent être beaucoup plus tardives jusqu’à 10 ans justifiant une surveillance prolongée (47). Près de la moitié des récidives sont asymptomatiques découvertes sur l’IRM de surveillance (56). Elles sont le plus souvent focales ou multifocales dans la SNC et dans moins de 10% des cas, purement spinales. Elles sont plus fréquentes dans les MB de groupe 4 (36).
A la récidive, la médiane de survie est inférieure à 3 ans (56). Le traitement à la récidive n’est pas standardisé et doit se faire si possible dans le cadre de protocoles. Il repose sur la chirurgie lorsqu’une exérèse satisfaisante semble possible. La réintervention aura également l’avantage de permettre une nouvelle étude histo-moléculaire et parfois de découvrir une tumeur d’une autre nature (tumeur radio induite) (5,41). Une ré-irradiation focale est parfois envisageable si la récidive est tardive (64).
Le rôle de la chimiothérapie est mal connu et palliative. Des molécules à faible toxicité comme le TEMOZOLOMIDE seul ou combiné au BEVACIZUMAB peuvent être proposées (65). Des protocoles beaucoup plus lourds associant chimiothérapie à haute dose avec greffe de moelle ont aussi été tentés mais avec une efficacité limitée (66). Des thérapies ciblées inhibitrices de la voie SHH sont actuellement testées dans les MB SHH mutés et font naître un certain espoir dans les récidives voire en première intention (67).
VIII. Conclusions
Le MB de l’adulte est une tumeur cérébrale maligne rare dont le pronostic est très satisfaisant dans certains sous-types moléculaires. La prise en charge s’inspire de celle de l’enfant même si des différences existent sur le plan clinique et biologique. Le traitement repose sur une exérèse chirurgicale le plus large possible suivi d’une irradiation cranio-spinale. Dans les MB classés à haut risque une chimiothérapie est systématiquement réalisée ; dans les MB à risque standard la chimiothérapie complémentaire est plus discutée mais actuellement volontiers proposée. Une surveillance prolongée clinique et par IRM est nécessaire pour détecter des toxicités liées au traitement et rechercher une dissémination lepto-méningée qui peut être tardive. Les progrès viendront d’une meilleure connaissance des différents sous types moléculaires, ce qui permettra de proposer dans le futur des thérapies ciblées.
Points forts
Les médulloblastomes sont des tumeurs cérébrales malignes rares chez l’adulte (1% des tumeurs intra-crâniennes) et sont différents des formes pédiatriques sur le plan clinique, moléculaire et pronostique.
La classification actuelle des médulloblastomes identifie 4 groupes histopathologiques (médulloblastomes classiques, desmoplasiques/nodulaires, anaplasiques/à grandes cellules, à nodularité eextensive) et 4 groupes moléculaires (WNT-activé, SHH-activé, groupe 3, groupe 4) qui ne sont pas strictement concordants.
La présentation clinique est dominée par une hypertension intra-crânienne, un syndrome cérébelleux.
Le bilan paraclinique comporte au minimum une IRM cérébrale, une IRM médullaire et une ponction lombaire, visant à caractériser la localisation exacte de la tumeur cérébelleuse, de rechercher une hydrocéphalie associée et une éventuellement dissémination lepto-méningée.
La prise en charge des médulloblastomes de l’adulte repose d’abord sur la chirurgie qui a pour objectifs de réaliser une exérèse maximale dont l’extension sera évaluée sur une IRM précoce (dans les 48 heures suivant la chirurgie) et de traiter une éventuelle hydrocéphalie. Pour les médulloblastomes à risque standard (résidu tumoral inférieur à 1,5 cm3, M0 (absence de métastase) ou M1 (présence de cellules dans le liquide cérébrospinal), type classique ou desmoplastique et de sous-type moléculaire WNT ou SHH), le traitement adjuvant repose sur la radiothérapie cranio-spinale souvent associée à une chimiothérapie. Pour les médulloblastomes à haut risque (résidu supérieur à 1,5cm3, M2, M3, M4, sous-type moléculaire non WNT ou non SHH ou SHH TP53 muté), une chimiothérapie sera systématiquement proposée en plus de la radiothérapie.
La surveillance post-thérapeutique repose sur l’IRM cérébrale, réalisée tous les 3 à 6 mois pendant 5 ans puis tous les ans pendant 10 ans complétée par une IRM médullaire pendant les 2 premières années. Ces imageries ont pour but de diagnostiquer les récidives qui surviennent généralement dans les 2 à 4 ans suivant la prise en charge initiale et de dépister d’éventuelles complications liées aux traitements, avec une attention particulière vis-à-vis des séquelles cognitives tardives liées à la radiothérapie qui sont fréquentes.
A la récidive, et après un nouveau bilan par imagerie la prise en charge repose sur la chirurgie lorsqu’elle est possible, la ré-irradiation lorsqu’elle est possible et la chimiothérapie.
A 5 ans, la survie globale varie de 82% à 100% pour les formes standards et de à 62% à 73% pour les formes à haut risque.
Réponses aux QCM
1. Réponses vraies : CD
A. Faux : Le médulloblastome est rare chez l’adulte
B. Faux : Les médulloblastomes de l’adulte sont souvent révélés par un syndrome cérébelleux à la différence de l’ enfant
C. Faux : une PL avant l’ IRM spinale peut induire des faux positifs
D. Vrai
E. Faux : Il s’agit des formes hémisphériques très latérales
2. Réponse vraie : E
A. Faux : Les groupes histologiques et moléculaires ne sont pas strictement concordants
B. Faux : Les médulloblastomes desmoplasiques/nodulaires présentent habituellement une activation de la voie SHH
C. Faux : Les médulloblastomes du groupe 3 sont de mauvais pronostic
D. Faux : Les médulloblastomes desmoplasiques/nodulaires et à nodularité extensive sont parfois associés au syndrome de Gorlin
E. Vrai
3. Réponses vraies : BCE
A. Faux : L’hydrocéphalie est habituellement obstructive et triventriculaire
B. Vrai : La prise de contraste peut être hétérogène (portions kystiques) ou homogène ou absente
C. Vrai : La restriction de diffusion s’observe au niveau des zones hypercellulaires
D. Faux : Le rCBV est habituellement peu ou pas augmenté
E. Vrai : La dissémination lepto-méningée peut en effet s’observer au niveau rachidien mais également au niveau encéphalique
4. Réponses vraies : BCDE
A. Faux : Absence de résidu tumoral, M1, type classique et sous-type WNT
B. Vrai : Résidu tumoral > 1,5 cm3
C. Vrai : Sous-type moléculaire SHH-activé et TP53 muté
D. Vrai : Métastase à distance
E. Vrai : Sous-type moélculaire = groupe 3
5. Réponses vraies : AE
A. Vrai
B. Faux : La chirurgie d’exérèse doit toujours viser à réaliser une exérèse maximale tout en limitant le risque fonctionnel.
C. Faux : La réalisation d’une IRM post-opératoire est recommandée dans les 48 heures suivant l’intervention afin d’évaluer la qualité de l’exérèse. Au-delà il y a des artefacts d’ interprétation
D. Faux : La réalisation d’un traitement adjuvant est systématique et sa nature dépend du risque lié aux caractéristiques tumorales
E. Vrai : A l’issue du traitement, la surveillance clinique et radiologique doit être poursuivie pendant au moins 10 ans afin de dépister les récidives tardives et les complications liées aux traitements.
Références essentielles
Franceschi E, Hofer S, Brandes AA, Frappaz D, Kortmann R-D, Bromberg J, et al. EANO-EURACAN clinical practice guideline for diagnosis, treatment, and follow-up of post-pubertal and adult patients with medulloblastoma. Lancet Oncol. déc 2019 ;20(12):e715‑28.
Il s’agit des recommandations de l’EANO (European Association of NeuroOncology) et de l’EURACAN (EUropean Rare CANcers), relatives au diagnostic et à la prise en charge des médulloblastomes de l’adulte.
Majd N, Penas-Prado M. Updates on Management of Adult Medulloblastoma. Curr Treat Options Oncol. 24 juin 2019 ;20(8):64.
Il s’agit d’une revue de la littérature consacrée à la prise en charge des médulloblastomes de l’adulte synthétisant les données des essais cliniques les plus récents ayant évalué le rôle des chimiothérapies classiques, des thérapies ciblées et de l’immunothérapie.
Eibl T, Hammer A, Yakubov E, Blechschmidt C, Kalisch A, Steiner H-H. Medulloblastoma in adults - reviewing the literature from a surgeon’s point of view. Aging (Albany NY). 26 janv 2021 ;13(2):3146‑60.
Il s’agit d’une revue de la littérature concernant les médulloblastomes de l’adulte, focalisée sur les aspects chirurgicaux.
Majd NK, Mastall M, Lin H, Dibaj SS, Hess KR, Yuan Y, et al. Clinical characterization of adult medulloblastoma and the effect of first-line therapies on outcome ; The MD Anderson Cancer Center experience. Neurooncol Adv. déc 2021 ;3(1).
Il s’agit d’une étude rétrospective ayant inclus 200 patients adultes fournissant une analyse pronostique détaillée.
Siegfried A, Delisle M-B. [Medulloblastoma. Pathology]. Neurochirurgie. févr 2021 ;67(1):28‑38.
Il s’agit d’une revue détaillée à propos de la classification histologique et moléculaire des médulloblastomes.
Références bibliographiques
1. Farwell JR, Dohrmann GJ, Flannery JT. Medulloblastoma in childhood : an epidemiological study. J Neurosurg. oct 1984 ;61(4):657‑64.
2. Smoll NR. Relative survival of childhood and adult medulloblastomas and primitive neuroectodermal tumors (PNETs). Cancer. 1 mars 2012 ;118(5):1313‑22.
3. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report : Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol. 1 oct 2018 ;20(suppl_4):iv1‑86.
4. Waszak SM, Northcott PA, Buchhalter I, Robinson GW, Sutter C, Groebner S, et al. Spectrum and prevalence of genetic predisposition in medulloblastoma : a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. juin 2018 ;19(6):785‑98.
5. Franceschi E, Hofer S, Brandes AA, Frappaz D, Kortmann R-D, Bromberg J, et al. EANO-EURACAN clinical practice guideline for diagnosis, treatment, and follow-up of post-pubertal and adult patients with medulloblastoma. Lancet Oncol. déc 2019 ;20(12):e715‑28.
6. Frappaz D, Faure-Conter C, Bonneville Levard A, Barritault M, Meyronet D, Sunyach M-P. Medulloblastomas in adolescents and adults - Can the pediatric experience be extrapolated ? Neurochirurgie. févr 2021 ;67(1):76‑82.
7. Kool M, Korshunov A, Pfister SM. Update on molecular and genetic alterations in adult medulloblastoma. Memo. sept 2012 ;5(3):228‑32.
8. Korshunov A, Remke M, Werft W, Benner A, Ryzhova M, Witt H, et al. Adult and pediatric medulloblastomas are genetically distinct and require different algorithms for molecular risk stratification. J Clin Oncol. 20 juin 2010 ;28(18):3054‑60.
9. Massimino M, Sunyach MP, Barretta F, Gandola L, Garegnani A, Pecori E, et al. Reduced-dose craniospinal irradiation is feasible for standard-risk adult medulloblastoma patients. J Neurooncol. juill 2020 ;148(3):619‑28.
10. Hau P, Frappaz D, Hovey E, McCabe MG, Pajtler KW, Wiestler B, et al. Development of Randomized Trials in Adults with Medulloblastoma-The Example of EORTC 1634-BTG/NOA-23. Cancers (Basel). 9 juill 2021 ;13(14):3451.
11. Siegfried A, Delisle M-B. [Medulloblastoma. Pathology]. Neurochirurgie. févr 2021 ;67(1):28‑38.
12. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System : a summary. Neuro-Oncology. 1 août 2021 ;23(8):1231‑51.
13. Pietsch T, Haberler C. Update on the integrated histopathological and genetic classification of medulloblastoma - a practical diagnostic guideline. Clin Neuropathol. déc 2016 ;35(6):344‑52.
14. Siegfried A, Bertozzi AI, Bourdeaut F, Sevely A, Loukh N, Grison C, et al. Clinical, pathological, and molecular data on desmoplastic/nodular medulloblastoma : case studies and a review of the literature. Clin Neuropathol. juin 2016 ;35(3):106‑13.
15. Gessi M, Goschzik T, Dörner E, Söldner K, Schupp C, Pietsch T. Medulloblastoma with extensive nodularity : a tumour exclusively of infancy ? Neuropathol Appl Neurobiol. avr 2017 ;43(3):267‑70.
16. Goschzik T, Zur Mühlen A, Kristiansen G, Haberler C, Stefanits H, Friedrich C, et al. Molecular stratification of medulloblastoma : comparison of histological and genetic methods to detect Wnt activated tumours. Neuropathol Appl Neurobiol. févr 2015 ;41(2):135‑44.
17. Pfaff E, Remke M, Sturm D, Benner A, Witt H, Milde T, et al. TP53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J Clin Oncol. 10 déc 2010 ;28(35):5188‑96.
18. Northcott PA, Jones DTW, Kool M, Robinson GW, Gilbertson RJ, Cho Y-J, et al. Medulloblastomics : the end of the beginning. Nat Rev Cancer. déc 2012 ;12(12):818‑34.
19. Zhao F, Ohgaki H, Xu L, Giangaspero F, Li C, Li P, et al. Molecular subgroups of adult medulloblastoma : a long-term single-institution study. Neuro Oncol. juill 2016 ;18(7):982‑90.
20. von Bueren AO, Friedrich C, von Hoff K, Kwiecien R, Müller K, Pietsch T, et al. Metastatic medulloblastoma in adults : outcome of patients treated according to the HIT2000 protocol. Eur J Cancer. nov 2015 ;51(16):2434‑43.
21. Majd NK, Mastall M, Lin H, Dibaj SS, Hess KR, Yuan Y, et al. Clinical characterization of adult medulloblastoma and the effect of first-line therapies on outcome ; The MD Anderson Cancer Center experience. Neurooncol Adv. déc 2021 ;3(1):vdab079.
22. Dirven L, Luerding R, Beier D, Bumes E, Reinert C, Seidel C, et al. Neurocognitive functioning and health-related quality of life in adult medulloblastoma patients : long-term outcomes of the NOA-07 study. J Neurooncol. mai 2020 ;148(1):117‑30.
23. Ma AK, Freedman I, Lee JH, Miyagishima D, Ahmed O, Yeung J. Tumor Location and Treatment Modality are Associated with Overall Survival in Adult Medulloblastoma. Cureus. 20 févr 2020 ;12(2):e7061.
24. Ang C, Hauerstock D, Guiot M-C, Kasymjanova G, Roberge D, Kavan P, et al. Characteristics and outcomes of medulloblastoma in adults. Pediatr Blood Cancer. nov 2008 ;51(5):603‑7.
25. Vinchon M, Leblond P. Medulloblastoma : Clinical presentation. Neurochirurgie. févr 2021 ;67(1):23‑7.
26. Beier D, Kocakaya S, Hau P, Beier CP. The Neuroradiological Spectra of Adult and Pediatric Medulloblastoma Differ : Results from a Literature-based Meta-analysis. Clin Neuroradiol. mars 2018 ;28(1):99‑107.
27. Dangouloff-Ros V, Varlet P, Levy R, Beccaria K, Puget S, Dufour C, et al. Imaging features of medulloblastoma : Conventional imaging, diffusion-weighted imaging, perfusion-weighted imaging, and spectroscopy : From general features to subtypes and characteristics. Neurochirurgie. févr 2021 ;67(1):6‑13.
28. Fruehwald-Pallamar J, Puchner SB, Rossi A, Garre ML, Cama A, Koelblinger C, et al. Magnetic resonance imaging spectrum of medulloblastoma. Neuroradiology. juin 2011 ;53(6):387‑96.
29. Koob M, Girard N. Cerebral tumors : specific features in children. Diagn Interv Imaging. oct 2014 ;95(10):965‑83.
30. Theillac M, Meyronet D, Savatovsky J, Makris N, Hannoun S, Grand S, et al. Dynamic susceptibility contrast perfusion imaging in biopsy-proved adult medulloblastoma. J Neuroradiol. oct 2016 ;43(5):317‑24.
31. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 23 déc 2010 ;468(7327):1095‑9.
32. Perreault S, Ramaswamy V, Achrol AS, Chao K, Liu TT, Shih D, et al. MRI surrogates for molecular subgroups of medulloblastoma. AJNR Am J Neuroradiol. juill 2014 ;35(7):1263‑9.
33. Łastowska M, Jurkiewicz E, Trubicka J, Daszkiewicz P, Drogosiewicz M, Malczyk K, et al. Contrast enhancement pattern predicts poor survival for patients with non-WNT/SHH medulloblastoma tumours. J Neurooncol. mai 2015 ;123(1):65‑73.
34. Eibl T, Hammer A, Yakubov E, Blechschmidt C, Kalisch A, Steiner H-H. Medulloblastoma in adults - reviewing the literature from a surgeon’s point of view. Aging (Albany NY). 26 janv 2021 ;13(2):3146‑60.
35. Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell. 12 juin 2017 ;31(6):737-754.e6.
36. Franceschi E, Seidel C, Sahm F, Pajtler KW, Hau P. How we treat medulloblastoma in adults. ESMO Open. août 2021 ;6(4):100173.
37. Spektor S, Fraifeld S, Margolin E, Saseedharan S, Eimerl D, Umansky F. Comparison of outcomes following complex posterior fossa surgery performed in the sitting versus lateral position. J Clin Neurosci. avr 2015 ;22(4):705‑12.
38. Rath GP, Bithal PK, Chaturvedi A, Dash HH. Complications related to positioning in posterior fossa craniectomy. J Clin Neurosci. juin 2007 ;14(6):520‑5.
39. Saladino A, Lamperti M, Mangraviti A, Legnani FG, Prada FU, Casali C, et al. The semisitting position : analysis of the risks and surgical outcomes in a contemporary series of 425 adult patients undergoing cranial surgery. J Neurosurg. oct 2017 ;127(4):867‑76.
40. Klein O, Boussard N, Guerbouz R, Helleringer M, Joud A, Puget S. Surgical approach to the posterior fossa in children, including anesthetic considerations and complications : The prone and the sitting position. Technical note. Neurochirurgie. févr 2021 ;67(1):46‑51.
41. Majd N, Penas-Prado M. Updates on Management of Adult Medulloblastoma. Curr Treat Options Oncol. 24 juin 2019 ;20(8):64.
42. Wibroe M, Rochat P, Juhler M. Cerebellar Mutism Syndrome and Other Complications After Surgery in the Posterior Fossa in Adults : A Prospective Study. World Neurosurg. févr 2018 ;110:e738‑46.
43. Chang CH, Housepian EM, Herbert C. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology. déc 1969 ;93(6):1351‑9.
44. Packer RJ, Goldwein J, Nicholson HS, Vezina LG, Allen JC, Ris MD, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy : A Children’s Cancer Group Study. J Clin Oncol. juill 1999 ;17(7):2127‑36.
45. Gilbertson R, Wickramasinghe C, Hernan R, Balaji V, Hunt D, Jones-Wallace D, et al. Clinical and molecular stratification of disease risk in medulloblastoma. Br J Cancer. 1 sept 2001 ;85(5):705‑12.
46. Franceschi E, Minichillo S, Mura A, Tosoni A, Mascarin M, Tomasello C, et al. Adjuvant chemotherapy in average-risk adult medulloblastoma patients improves survival : a long term study. BMC Cancer. 12 août 2020 ;20(1):755.
47. Brandes AA, Franceschi E, Tosoni A, Blatt V, Ermani M. Long-term results of a prospective study on the treatment of medulloblastoma in adults. Cancer. 1 nov 2007 ;110(9):2035‑41.
48. Liu I-C, Holtzman AL, Rotondo RL, Indelicato DJ, Gururangan S, Cavaliere R, et al. Proton therapy for adult medulloblastoma : Acute toxicity and disease control outcomes. J Neurooncol. juill 2021 ;153(3):467‑76.
49. Brown AP, Barney CL, Grosshans DR, McAleer MF, de Groot JF, Puduvalli VK, et al. Proton beam craniospinal irradiation reduces acute toxicity for adults with medulloblastoma. Int J Radiat Oncol Biol Phys. 1 juin 2013 ;86(2):277‑84.
50. Padovani L, Sunyach M-P, Perol D, Mercier C, Alapetite C, Haie-Meder C, et al. Common strategy for adult and pediatric medulloblastoma : a multicenter series of 253 adults. Int J Radiat Oncol Biol Phys. 1 juin 2007 ;68(2):433‑40.
51. Kocakaya S, Beier CP, Beier D. Chemotherapy increases long-term survival in patients with adult medulloblastoma—a literature-based meta-analysis. Neuro Oncol. mars 2016 ;18(3):408‑16.
52. Kann BH, Lester-Coll NH, Park HS, Yeboa DN, Kelly JR, Baehring JM, et al. Adjuvant chemotherapy and overall survival in adult medulloblastoma. Neuro Oncol. 1 févr 2017 ;19(2):259‑69.
53. Franceschi E, Bartolotti M, Paccapelo A, Marucci G, Agati R, Volpin L, et al. Adjuvant chemotherapy in adult medulloblastoma : is it an option for average-risk patients ? J Neurooncol. juin 2016 ;128(2):235‑40.
54. Lai R. Survival of patients with adult medulloblastoma : a population-based study. Cancer. 1 avr 2008 ;112(7):1568‑74.
55. Curran EK, Sainani KL, Le GM, Propp JM, Fisher PG. Gender affects survival for medulloblastoma only in older children and adults : a study from the Surveillance Epidemiology and End Results Registry. Pediatr Blood Cancer. janv 2009 ;52(1):60‑4.
56. Riffaud L, Saikali S, Leray E, Hamlat A, Haegelen C, Vauleon E, et al. Survival and prognostic factors in a series of adults with medulloblastomas. J Neurosurg. sept 2009 ;111(3):478‑87.
57. Thompson EM, Hielscher T, Bouffet E, Remke M, Luu B, Gururangan S, et al. Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup : a retrospective integrated clinical and molecular analysis. Lancet Oncol. avr 2016 ;17(4):484‑95.
58. Wong GC-H, Li KK-W, Wang W-W, Liu AP-Y, Huang QJ, Chan AK-Y, et al. Clinical and mutational profiles of adult medulloblastoma groups. Acta Neuropathol Commun. 10 nov 2020 ;8(1):191.
59. Northcott PA, Hielscher T, Dubuc A, Mack S, Shih D, Remke M, et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. août 2011 ;122(2):231‑40.
60. Kool M, Korshunov A, Remke M, Jones DTW, Schlanstein M, Northcott PA, et al. Molecular subgroups of medulloblastoma : an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. avr 2012 ;123(4):473‑84.
61. Abacioglu U, Uzel O, Sengoz M, Turkan S, Ober A. Medulloblastoma in adults : treatment results and prognostic factors. Int J Radiat Oncol Biol Phys. 1 nov 2002 ;54(3):855‑60.
62. Friedrich C, von Bueren AO, von Hoff K, Kwiecien R, Pietsch T, Warmuth-Metz M, et al. Treatment of adult nonmetastatic medulloblastoma patients according to the paediatric HIT 2000 protocol : a prospective observational multicentre study. Eur J Cancer. mars 2013 ;49(4):893‑903.
63. Harrison RA, Kesler SR, Johnson JM, Penas-Prado M, Sullaway CM, Wefel JS. Neurocognitive dysfunction in adult cerebellar medulloblastoma. Psychooncology. janv 2019 ;28(1):131‑8.
64. Milker-Zabel S, Zabel A, Thilmann C, Zuna I, Hoess A, Wannenmacher M, et al. Results of three-dimensional stereotactically-guided radiotherapy in recurrent medulloblastoma. J Neurooncol. déc 2002 ;60(3):227‑33.
65. Levy AS, Krailo M, Chi S, Villaluna D, Springer L, Williams-Hughes C, et al. Temozolomide with irinotecan versus temozolomide, irinotecan plus bevacizumab for recurrent medulloblastoma of childhood : Report of a COG randomized Phase II screening trial. Pediatr Blood Cancer. août 2021 ;68(8):e29031.
66. Grill J, Geoerger B, Gesner L, Perek D, Leblond P, Cañete A, et al. Phase II study of irinotecan in combination with temozolomide (TEMIRI) in children with recurrent or refractory medulloblastoma : a joint ITCC and SIOPE brain tumor study. Neuro Oncol. sept 2013 ;15(9):1236‑43.
67. Menyhárt O, Győrffy B. Molecular stratifications, biomarker candidates and new therapeutic options in current medulloblastoma treatment approaches. Cancer Metastasis Rev. mars 2020 ;39(1):211‑33.