Adénomes hypophysaires
Rédigé en février 2015 par Jeudi 19 février 2015, par Alexandre Vasiljevic , Emmanuel Jouanneau, Gérald Raverot , Gilles Brassier, Moncef Berhouma , Pierre-Louis Henaux , Timothée Jacquesson , Véronique Favrel, Véronique Lapras
1. Définition
Les adénomes hypophysaires sont des tumeurs développées à partir des cellules de l’antéhypophyse.
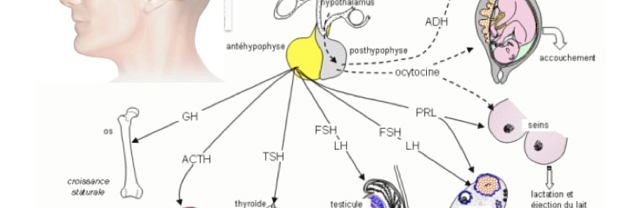
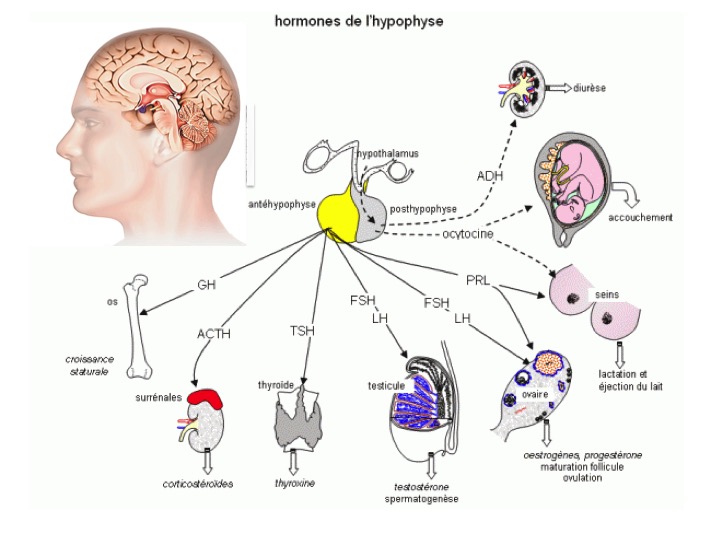
L’hypophyse (figure 1) est une glande d’environ 10 mm composée de 2 parties : l’antéhypophyse ou adénohypophyse qui sécrètent les hormones ACTH (Adrenocorticotrophin Hormon), GH (Growth Hormon), TSH (Thyroid Stimuling Hormon), FSH (Follicle Stimuling Hormon), LH (Lutropin Hormon), PRL (Prolactin) sous l’influence de l’hypothalamus, et la post hypophyse ou neurohypophyse qui sécrètent l’ocytocine (action utérine) et la vasopressine ou ADH (Anti Diuretic Hormon, action antidiurétique) qui contrôlent le volume de l’excrétion urinaire par rapport aux boissons.
Ces tumeurs sont le plus souvent bénignes et les cellules tumorales conservant une partie de leurs propriétés peuvent sécréter les hormones précitées.
2. Historique
L’anatomie de l’hypophyse est connue depuis l’antiquité gréco-romaine avec notamment les travaux d’Hippocrate et de Galien 29,43. Ce dernier en fait une voie d’ « évacuation des déchets de l’encéphale », notion qui sera enseignée pendant des millénaires aux étudiants en médecine. Un début de remise en cause des notions introduites par Galien débutera avec les travaux de Vespasien 73 pendant la Renaissance mais la relation entre maladies et tumeurs hypophysaires ne commencera réellement qu’avec la méthode anatomo-clinique au 17ième siècle. Jean Louis Petit (1674-1750) démontre ainsi que le développement d’une tumeur de l’hypophyse peut causer une cécité 43. Pierre Marie (1853-1940), en 1886, fait le lien entre les symptômes de l’acromégalie et une tumeur de l’hypophyse 46. L’hypopituitarisme est relié à un dysfonctionnement de l’hypophyse en 1901 par A Froelich (1871-1953) et H Cushing dans son ouvrage « The Pituitary Body and its Disorders » décrira finalement les différents tableaux cliniques d’hypo ou d’hypersécrétion connus de nos jours 21,43.
3. Epidémiologie et classifications des adénomes hypophysaires
Les adénomes hypophysaires représentent entre 10 et 20% des tumeurs intracrâniennes de l’adulte. Ces tumeurs ont une prévalence de 78 à 94/100 000 habitants et une incidence de 4/100 000 habitants avec une tendance à l’augmentation des découvertes fortuites, « incidentalomes » 37. Il s’agit d’une tumeur rare (2,7% des tumeurs supratentorielles) 39 chez l’enfant dont la tumeur de la région hypophysaire est le plus souvent un craniopharyngiome.
Depuis l’avènement de l’immunohistochimie, plusieurs sous-types histologiques correspondants aux différents tableaux cliniques sont individualisés.
Il est ainsi décrit des adénomes sécrétants ou fonctionnels :
– L’adénome à prolactine (PRL) est le plus fréquent (60% de l’ensemble des adénomes, ratio femme-homme : 10/1, une prévalence de 440/100000 et un âge moyen au diagnostic de 32 ans) 37.
– L’adénome somatotrope sécrétant l’hormone de croissance (GH) représente environ 13 % de l’ensemble des adénomes avec une prévalence estimée à 40 à 130/million ; l’âge au diagnostic est de 47 ans 37 avec une variante fréquente l’adénome somatoprolactinique, sécrétant de la PRL et de la GH (50% des adénomes somatotropes).
– L’adénome corticotrope a une prévalence de 0,17/100000 37. Ces adénomes majoritairement de petite taille (<10mm) ou « micro-adénomes » sécrètent de l’ACTH et représentent 6% de l’ensemble des adénomes. Les macroadénomes sont moins fréquents.
– L’adénome thyréotrope secrétant la TSH est rare (<1% de l’ensemble des adénomes) avec une prévalence de 0,15/2,8 millions 53. Il s’accompagne le plus souvent d’une hyperthyroïdie, mais il peut être silencieux et plurisécrétant (TSH-GH et TSH-PRL).
A côté de ces adénomes dit sécrétants sont décrits des adénomes non sécrétants ou non fonctionnels (30% des adénomes hypophysaires). Ce dernier terme « non fonctionnel » doit être préféré, car dans environ un tiers des cas 22,23,35, il existe une expression hormonale, sans traduction clinique, mais pouvant servir de marqueur biologique.
– Ces tumeurs sont en majorité des adénomes gonadotropes, issus des cellules gonadotropes sécrétant FSH, LH ou la sous-unité α (ces hormones « gonadotrophines », glycoprotidiques étant composées de 2 sous-unités α et β).
– Les adénomes silencieux sont moins fréquents et habituellement considérés comme agressifs. Il s’agit d’adénomes se révélant par un syndrome tumoral sans tableau clinique spécifique d’hypersécrétion, mais qui en immunocytochimie sont positifs pour l’ACTH, la GH, ou la TSH voire plurihormonaux (GH et PRL ou TSH et GH). Biologiquement, soit ils ne sécrètent pas d’hormone, soit plus fréquemment, ils sécrètent une pro-hormone inactive expliquant l’absence de signes cliniques ou une hormone en faible quantité, mais dosable (hypersécrétion d’ACTH sans hypercorticisme pour l’exemple le plus fréquent).
– Les adénomes immunonégatifs ne réagissant avec aucun anticorps sont actuellement très rares (<5%). Il s’agirait d’une forme indifférenciée d’adénomes gonadotropes.
Les adénomes hypophysaires sont considérés comme des tumeurs bénignes mais il existe de véritables adénomes agressifs ou atypiques et carcinomes. La fréquence des adénomes agressifs est encore méconnue, 15% de grade 2b dans l’étude de J Trouillas et al 75 ainsi que dans la série de Laws 80. Seules 0,2% des tumeurs présentant des métastases au cours de l’évolution sont considérées comme des carcinomes : sécrétants pour la plupart - 36% de PRL, 30% d’ACTH - et non sécrétants dans uniquement 23%. Ces métastases jamais inaugurales surviennent 5 à 16 ans après le début de l’histoire clinique et concernent soit le système nerveux avec une dissémination par le liquide cérébro-spinal (LCS) soit systémiques pulmonaires, osseuses, hépatiques voire dans les ganglions lymphatiques (dissémination hématogène) 26. La survie des patients est alors de 4 ans.
Selon la WHO (World Health Organization) classification de l’OMS [1], ces tumeurs sont classées en 3 types : les adénomes bénins, « atypiques » (exprimant fortement la p53, de nombreuses mitoses, sans valeur seuil pour ces 2 marqueurs et un KI-67>3%) et les carcinomes (avec des métastases cérébro-spinales ou systémiques). Cette classification n’a pas de valeur pronostique, car ne s’accompagnant pas de corrélations anatomo-cliniques.
La deuxième classification plus récente est issue d’un travail français, multidisciplinaire et multicentrique dans le cadre d’une étude cas témoin rétrospective portant sur 410 patients opérés 75. Dans cette étude, l’invasion a été définie par un envahissement radiologique (IRM) et/ou histologique du sinus sphénoïdal - avec franchissement de la dure-mère et de l’os et surtout du sinus caverneux 20,38. La prolifération a été définie à l’histologie par la présence de 2 des trois marqueurs suivants : des mitoses (n>2/10 champs au 40), un index de KI-67 > 3% et une p53 positive (>10 noyaux/10 champs au 40). En fonction de l’invasion et de la prolifération, 5 grades ont été définis (figure 2) : grade 1a non invasif non proliférant, grade 1b non invasif mais proliférant, grade 2a invasif non proliférant, grade 2b invasif et proliférant, grade 3 métastatique. Cette classification s’est avérée capable de prédire la guérison et la récidive/progression à 8 ans quelque soit le sous type histologique. Elle individualise des tumeurs de grade 2b, à fort risque de résidu tumoral (25 fois supérieur au grade 1a) ou de récidive/progression (12 fois supérieur au grade 1a). L’objectif de ces classifications est de permettre au clinicien de s’appuyer sur des données objectives pour prendre des décisions de traitement complémentaire en cas de résidu post-chirurgical.

++++
4. Diagnostic
Le diagnostic est porté sur des symptômes cliniques qui motiveront alors un complément de bilan, sur une imagerie (découverte fortuite) ou sur des symptômes ophtalmologiques.
4.1 Eléments cliniques
Le prolactinome
Pour plus de détails, le lecteur pourra se référer aux articles de consensus suivants : Casanueva clin endocrinol 2006, Brue Ann Endo 2007, Melmed JCEM2011 [2].
Deux tableaux s’opposent :
– Le classique microadénome (<10 mm) ou « petit » macroprolactinome de la jeune femme responsable d’un syndrome d’aménorrhée (parfois spanio- ou oligoménorrhée)-galactorrhée (spontanée ou provoquée) avec baisse de la libido et stérilité, céphalées et prise de poids. Il s’agit d’une tumeur bien limitée et rarement invasive. Le problème est la restauration de la fertilité.
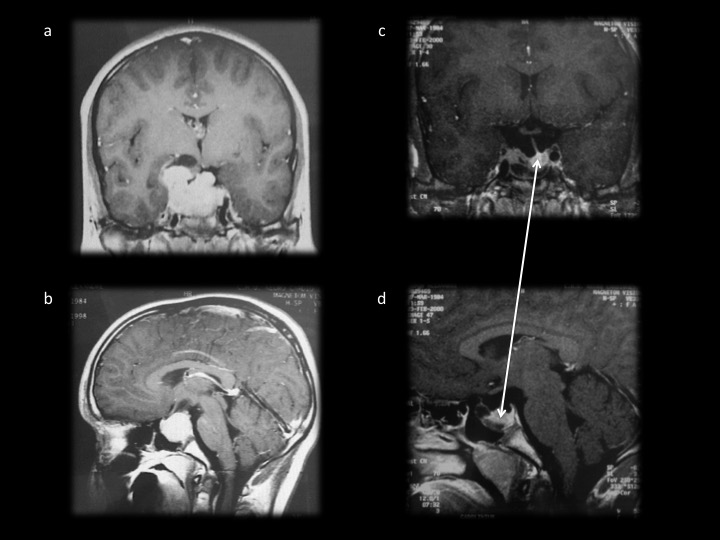
– Le prolactinome de l’homme de révélation plus tardive vers la cinquantaine par un syndrome tumoral avec troubles visuels (hémianopsie bitemporale pathognomonique). La baisse de la libido avec dysérection est la règle, mais n’amène à consulter que tardivement. Une gynécomastie peut exister mais la galactorrhée est rare. Les tumeurs de l’homme sont des macroadénomes, non en raison de leur révélation tardive mais de leurs caractères plus agressifs [3]. L’IRM montre alors un macroadénome parfois très volumineux (figure 3).
L’adénome somatotrope
Pour plus de détails, le lecteur peut se référer aux consensus français (Chanson Ann Endo 2009) et américain (Melmed JCEM 2009) sur l’acromégalie [4].
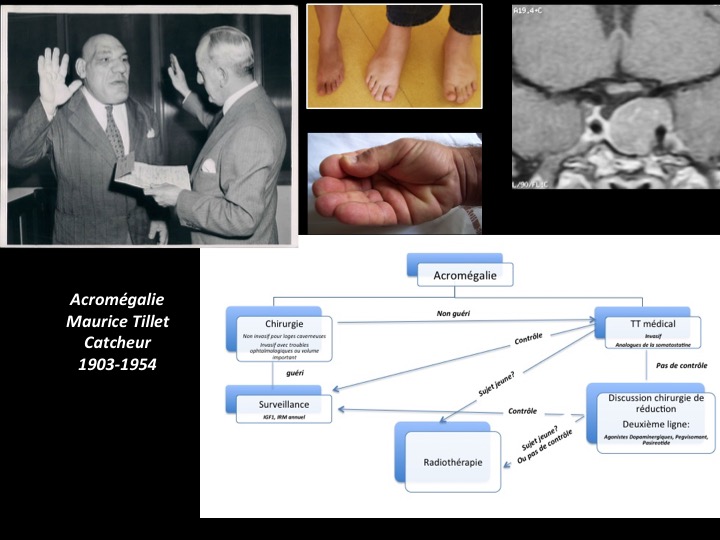
Sur le plan clinique, l’acromégalie se traduit par le classique syndrome dysmorphique associant augmentation de la pyramide nasale, des reliefs osseux notamment des arcades sourcilières, un prognathisme, une augmentation de la taille des mains et des pieds (figure 4). Le syndrome du canal carpien peut être inaugural. Les céphalées sont fréquentes, de même que les sueurs nocturnes malodorantes et très pénibles. Le syndrome d’apnée du sommeil (SAS) est la règle et doit être dépisté. Il peut être associé à une fatigue chronique. Une réaction dépressive est également fréquente. La morbidité de cette maladie est d’ordre cardiologique (HTA et une myocardiopathie hypertrophique), endocrinienne (intolérance au glucose ou un diabète) et rhumatologique touchant les grosses articulations (épaule, poignet, genou, hanche) et le rachis. Enfin un goitre est fréquent, ainsi que des polypes coliques qui justifient une coloscopie systématique dans le bilan initial. Il faut en effet souligner l’augmentation d’incidence des cancers coliques, mais aussi thyroïdiens dans l’acromégalie.
L’adénome corticotrope
Nous traiterons la maladie de Cushing avec un hypercorticisme ACTH dépendant dû à un adénome hypophysaire et non le syndrome de Cushing, avec un hypercorticisme, ACTH dépendant ou non. Ce paragraphe résume des articles de consensus récemment publiés (rapport HAS, Biller JCEM 2008) [5].
Les signes cliniques (figure 5) témoignent d’un hypercatabolisme (cutanée : vergetures larges et pourpres, ecchymoses, fragilité vasculaire ; musculaire avec amyotrophie proximale ; osseuse : ostéoporose) avec une répartition facio-tronculaire des graisses (obésité androïde avec bosse de bison ou Buffalo Neck, à la base du cou).
Chez l’enfant il existe un ralentissement de la courbe de croissance, avec une prise de poids paradoxale. Les morbidités sont sévères avec HTA, diabète, ostéoporose et une immunodépression sévère en plus du préjudice esthétique. Le retentissement psychologique voire psychiatrique est fréquent.
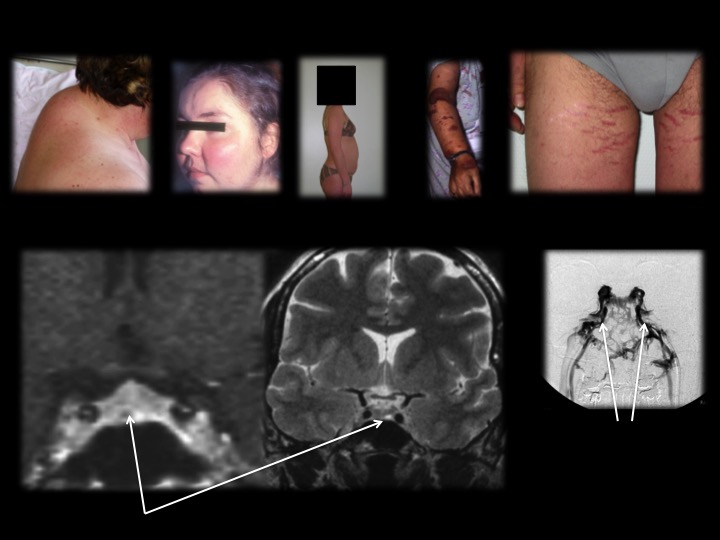
L’adénome thyréotrope
Les adénomes thyréotropes (sécrétant en excès de la TSH) sont rares représentant moins de 1% des adénomes hypophysaires.
Le tableau clinique est celui d’une thyréotoxicose avec nervosité, perte de poids et troubles cardiovasculaires (HTA, palpitations, troubles du rythme) associés à un syndrome tumoral (céphalées, troubles visuels) dans environ un quart des cas. L’aménorrhée est fréquente (25%) de même que la présence d’un goitre avec un pourcentage similaire [6].
Les adénomes hypophysaires non sécrétants ou non fonctionnels (gonadotropes et « silencieux »
Le consensus de la société Française d’Endocrinologie est à paraître dans les annales d’endocrinologie et un chapitre de l’EMC est consacré à ce type de tumeurs (Raverot et al. EMC 2014) [7].
Pour la plupart, ces adénomes hypophysaires ne secrètent pas d’hormones actives et se révèlent par un syndrome tumoral ou sont découvert de façon fortuite. En effet, la sécrétion de FSH, LH ou de sous-unité α (ces hormones « gonadotrophines », glycoprotidiques étant composées de 2 sous-unités α et β) est exceptionnellement responsables d’un tableau d’hyperstimulation ovarienne (kystes) ou d’hypertrophie testiculaire.
Sur le plan clinique, le mode de révélation se fait majoritairement sur des troubles visuels (l’hémianopsie bitemporale pathognomonique souvent asymétrique mais fréquemment associée à une baisse d’acuité visuelle unie ou bilatérale), une insuffisance antéhypophysaire notamment du secteur gonadotrope, fréquente (l’insuffisance posthypophysaire est exceptionnelle et doit faire remettre en question le diagnostic d’adénome) et des céphalées, sans caractéristiques particulières.
Les cas d’incidentalome sont en augmentation de fréquence.
Les adénomes hypophysaires atypiques et les carcinomes
Il n’y a pas de particularité clinique de ces adénomes en dehors leur caractère récidivant et résistant aux traitements classiques.
Apoplexie hypophysaire
L’apoplexie hypophysaire correspond à un syndrome clinique associant des céphalées brutales (véritable diagnostic différentiel des hémorragies sous-arachnoïdiennes), des troubles ophtalmologiques plus ou moins sévères (baisse acuité visuelle BAV, altération du champ visuel CV, paralysie oculomotrice) et/ou une insuffisance anté et post-hypophysaire avec hyponatrémie. Le contexte est donc celui de l’urgence parfois difficile à comprendre.
La physiopathologie est soit un infarctus, soit une hémorragie intra-adénomateuse qui augmentent brutalement le volume et la pression intra-sellaire.
Il s’agit soit d’un événement inaugural révélateur de la tumeur soit d’un événement compliquant l’histoire d’un adénome hypophysaire. L’incidence de l’apoplexie est faible (2 à 7% de l’ensemble des adénomes). Les facteurs favorisants retrouvés dans la littérature sont 60 :
– des risques cardiovasculaires : HTA, chirurgie cardiaque coronariennes notamment (rôle des variations pressionnels et des traitements anticoagulants)
– iatrogènes : anti-agrégants, anti-coagulants, tests hormonaux dynamiques lors de bilan pour adénome hypophysaire (GHRH, CRH, TSH), traitement par agonistes dopaminergiques, ou oestrogéniques
– la grossesse
– un traumatisme crânien
– une radiothérapie hypophysaire ?
La survenue est plus fréquente vers la 5ème ou 6ème décennie, avec une légère prédominance masculine.
++++
4.2 Eléments endocriniens
Il conviendra de « bilanter » l’hypersécrétion, les conséquences et dépister une éventuelle insuffisance antéhypophysaire.
Le prolactinome
Devant une hyperprolactinémie, avant de penser à une tumeur, on éliminera une grossesse (cause d’aménorrhée la plus fréquente chez la femme en période d’activité génitale) et une cause iatrogène d’hyperprolactinémie (psychotropes, anti-émétiques..) ainsi qu’une « big ou big-big PRL » (formes moléculaires).
Sur le plan biologique, devant un adénome et notamment un macroadénome, un dosage de la prolactinémie (PRL) est fondamental.
Le taux de PRL est corrélé au volume de l’adénome à savoir que pour un microadénome, la PRL se situera au environ de 150 μg/l alors qu’un macroadénome dépasse fréquemment les 1000 ou 2000 μg/l. Il conviendra aussi de se méfier d’un artefact de laboratoire ou « effet crochet ». Ainsi, un excès d’antigène par rapport aux anticorps peut conduire à une sous-estimation du dosage final avec un rendu de taux faible ou normal. Ce piège classique en technique de dosage IRMA (radioimmunometric assay) doit faire pratiquer des dilutions devant tout macroadénome avant conclusion définitive.
L’hyperPRL de déconnection (macroadénome comprimant la tige pituitaire empêchant le passage de la Dopamine inhibant la sécrétion de PRL) reste modérée (2 à 3 fois la norme et jamais au delà de 200μg/l). Des tests dynamiques (dosage de PRL après injection de TRH, L-DOPA, Domperidone, Metoclopramide) existent pour différencier hyperPRL de déconnection et hyperPRL tumorale (absence de réponse si adénome) mais leur valeur discriminante est discutée [8].
Une hyperPRL sans signe clinique doit faire penser à une macroprolactinémie, Big ou Big-Big prolactine (formes dimèriques ou polymériques ou agrégat résultant d’anticorps antiPRL) et justifiera une recherche spécifique [9].
Un bilan hypophysaire complet sera réalisé qui montrera un hypogonadisme souvent réactionnel à la seule hyperprolactinémie mais parfois secondaire au développement tumoral. Une co-sécrétion à GH peut être recherchée notamment en cas de macroadénome ou de signes cliniques évocateurs.
L’adénome somatotrope
Sur le plan biologique, un dosage de GH et d’IGF-1 est préconisé. Si le dosage de GH est inférieur à <0,4 μg/l (1,2 mUI/L) et le taux d’IGF-1 normal, le diagnostic est éliminé. Un dosage d’IGF-1 élevé est insuffisant pour poser le diagnostic d’acromégalie mais toute suspicion nécessite une consultation auprès d’un endocrinologue dans un centre référent pour les pathologies hypophysaires. Une hyperglycémie provoquée orale (HGPO) affirmera le diagnostic et dépistera une intolérance au glucose. L’absence de freinage du taux de GH confirme le diagnostic d’acromégalie (Réponse normale : GH < 0,3μg/l ou 0,9 mUI/L au nadir de l’HGPO). Un bilan endocrinien à la recherche d’une co-sécrétion de PRL ou de la sous-unité α des gonadotrophines ou d’un déficit hypophysaire doit être réalisé.
Le bilan biologique s’attachera à faire aussi une recherche des complications notamment un diabète avec une glycémie à jeun et une Hba1c.
Un dosage de GH-RH sera demandé si aucune tumeur hypophysaire n’est retrouvée dans l’hypothèse d’une exceptionnelle tumeur ectopique.
D’autres explorations sont nécessaires dans le cadre d’une acromégalie au diagnostic. Une consultation cardiologique (prise tensionnelle voire holter tensionnel, ECG et échocardiographie et dépistage d’un SAS), une colonoscopie quelque soit l’âge au diagnostic, une consultation rhumatologique auprès d’un praticien sensibilisé à la pathologie : radiographies à la demande selon les plaintes du patient, bilan phosphocalcique, ostéodensitométrie, une échographie abdominale et rénale (recherche de lithiases avant traitement médical), une échographie thyroïdienne compléteront le bilan.
Un questionnaire de qualité de vie spécifique (acroQOL) est disponible.
L’adénome corticotrope
Le diagnostic biologique est assuré par les dosages statiques : cycle nycthéméral d’ACTH-cortisol, cortisol salivaire à minuit, cortisoluries des 24h et par des tests dynamiques : freinage minute (1 mg de dexaméthasone à 23h) avec dosage du cortisol plasmatique à 8h le lendemain (normalité : cortisolémie freinée < 18 ng/ml ou 50 nmol/l) ou faible (dexaméthasone 0,5 mg/6h pendant deux jours), fort (2 mg per os toutes les 6 heures pendant deux jours, ou de 8 mg per os en une seule prise à minuit) avec réponse évaluée sur la cortisolurie le deuxième jour du test ou sur le cortisol plasmatique et éventuellement salivaire en fin de test et de stimulation : 1 /Test au CRH (100μg chez l’adulte ) avec réponse jugée sur l’ACTH-cortisol plasmatique ou cortisol urinaire. Une réponse positive franche est en faveur d’une origine hypophysaire et 2/ test à la desmopressine (administration en intraveineux de 10μg) avec les mêmes critères de jugement. Une stimulation paradoxale est en faveur d’une maladie de Cushing mais l’intérêt de ce test est limité par le fort pourcentage de tumeurs ectopiques répondant à la desmopressine.
En effet, le diagnostic différentiel d’un cushing ACTH dépendant par microadénome est un Cushing paranéoplasique. Le tableau clinique est en principe plus explosif et le profil des tests dynamiques différents (pas de freinage - absence de réponse au Minirin - desmopressine) mais parfois la recherche d’une tumeur ectopique (neuroendocrines pulmonaires souvent) s’impose (scanner thoraco-abdominal, cathéterisme des sinus pétreux, Tomographie par émission à Positron).
Le bilan biologique s’attachera à faire aussi un bilan hypophysaire complet et une recherche des complications notamment avec une glycémie à jeun et une Hba1c et un bilan lipidique.
L’adénome thyréotrope
Le tableau biologique est celui d’une élévation de T3 et T4 avec TSH normale ou élevée.
Le diagnostic différentiel est celui d’un syndrome de résistance périphérique aux hormones thyroïdiennes. Le diagnostic est assuré par :
– les tests dynamiques : il faut conserver en mémoire que l’adénome s’autonomise des voies de contrôles normales (test au TRH : stimulation moindre avec élévation de la TSH < à 200% ; test de suppression à la T3 : peu ou pas de diminution dans le cas d’adénome). Par contre, l’adénome répond de part ces récepteurs à la somatostatine, test à l’octréotide : diminution des taux de TSH.
– l’augmentation de la sous-unité alpha orientant vers l’adénome hypophysaire
– L’étude des récepteurs aux hormones thyroïdiennes (recherche de la mutation béta du gène TR) et une histoire familiale en faveur d’un syndrome de résistance aux hormones thyroïdiennes permettent de faire le diagnostic [10].
Un bilan hypophysaire complet est nécessaire tant pour faire le point sur les fonctions anté et post-hypophyaires que pour rechercher des co-sécrétions (alpha sous-unité, GH : un tiers de co-sécrétion TSH-GH).
Les adénomes hypophysaires non sécrétants ou non Fonctionnels
Un bilan endocrinien avec PRL (lors d’un volume important avec PRL normale se méfier d’un effet crochet et demander une dilution) et une exploration complète des fonctions hypophysaires à la recherche d’une insuffisance est la règle. L’insuffisance gonadotrope de même qu’une insuffisance partielle autre sont des situations fréquentes (un tiers des cas) alors que l’insuffisance antéhypophysaire totale est rare 51.
Les adénomes atypiques et carcinomes hypophysaires
Il n’y a pas de spécificité de ce type d’adénome et les fonctions hypophysaires feront l’objet d’une étude biologique classique.
L’apoplexie pituitaire
Le contexte est celui de l’urgence et le bilan doit comporter un bilan biologique avec un ionogramme sanguin avec créatinémie, recherche d’une hyponatrémie (fréquente par SIAH ou hypocorticisme), un bilan de coagulation avec NFP ainsi qu’un bilan endocrinien (PRL, cortisol, T3-T4, TSH, FSH, LH, testostérone, IGF-1). L’insuffisance antéhypophysaire est fréquente (entre 50 et 70%) [11].
Un bilan ophtalmologique (AV, CV, FO, Lancaster) ainsi qu’une IRM (le scanner cérébral est souvent le premier examen réalisé, mais l’IRM est la référence comme dans toute pathologie hypophysaire) devra être aussi demandé.
++++
4.3 Bilan ophtalmologique
Un bilan ophtalmologique sera réalisé dès lors que l’adénome dépasse la loge sellaire avec une extension suprasellaire au contact du chiasma. Il devra comporter une étude de l’acuité visuelle, du champ visuel automatisé, un fond d’œil (recherche d’une pâleur ou d’une atrophie papillaire de valeur pronostique). L’OCT (Tomographie par Cohérence Optique) est intéressante pour rechercher une altération du nerf optique de valeur pronostique 30,32. Un test de Lancaster sera demandé fonction de la clinique.
4.4 Eléments d’imagerie
Bilan radiologique de référence pour les adénomes hypophysaires
L’anatomie nasale (figure 6 a, b, c) à la recherche d’une déviation septale, d’une épine nasale, de mégacornets ou de concha Bullosa (pneumatisation du cornet moyen) doit être particulièrement étudiée pour prévoir la voie d’abord ce qui peut être fait par un scanner du massif facial mais l’IRM peut être suffisante. La pneumatisation et la septation du sinus sphénoïdal feront aussi l’objet d’une attention particulière. Une absence de sinus sphénoïdal (sinus Concha) ne contre-indique pas un abord trans-sphénoïdal mais le chirurgien devra se créer par fraisage un néo-sinus ce qui requière de l’expérience. La neuronavigation ou imagerie per-opératoire sont alors particulièrement utiles.
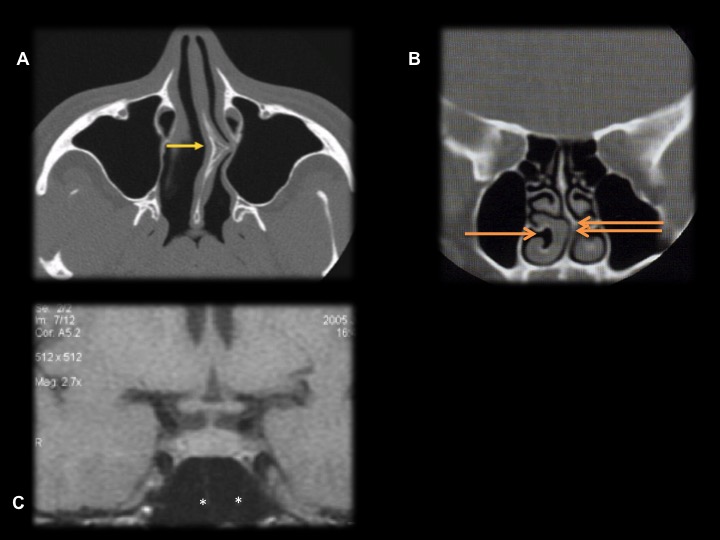
L’imagerie de référence pour l’hypophyse est l’IRM avec les séquences suivantes : coupes de 2 à 3 mm coronales et sagittales avec séquence T1 sans et avec injection de gadolinium et T2 coronales en écho de spin (figure 7).
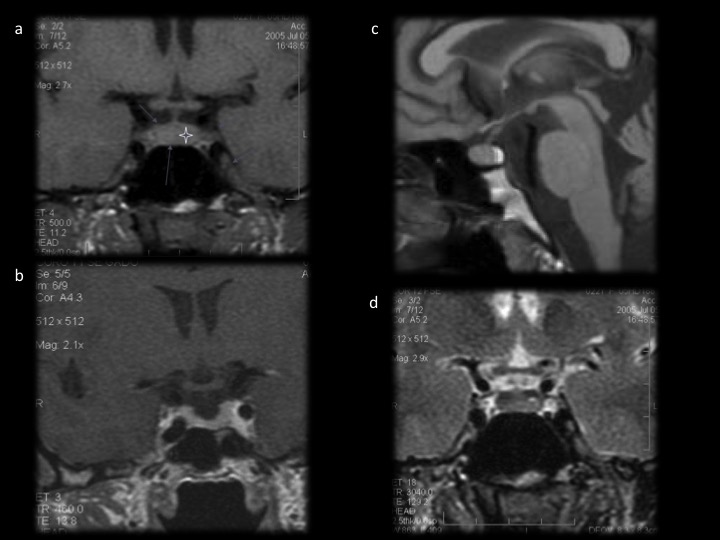
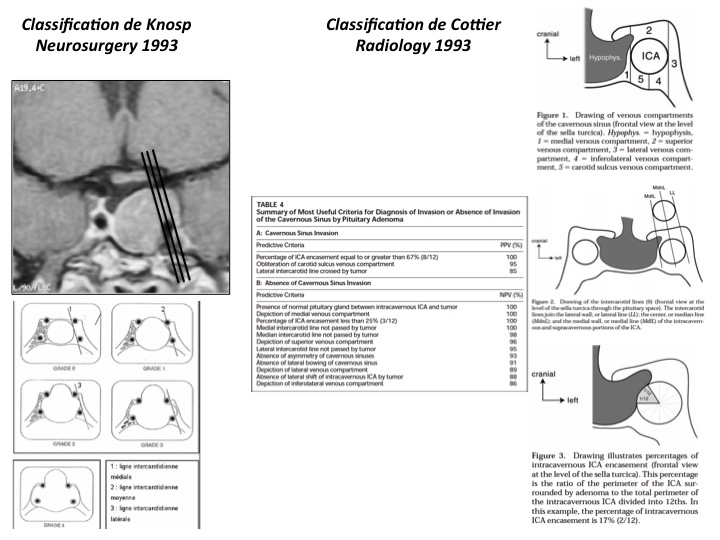
Une IRM de neuronavigation (séquences T1 gadolinium en 3D) est souhaitable dans l’hypothèse d’une chirurgie. Une attention toute particulière serait prêtée au dépistage d’un éventuel envahissement de la loge caverneuse. Deux classifications (Knosp, Cottier) sont en vigueur mais l’envahissement toujours difficile à affirmer en dehors des cas d’englobement de l’artère carotide interne intracaverneuse (figure 8).
L’hypophyse résiduelle peut souvent, même dans les cas de macroadénome, être visualisée sous forme d’un fin hypersignal au pôle supérieur de l’adénome (figure 9).
L’évaluation de la consistance est aussi intéressante pour le chirurgien, une tumeur ferme sera beaucoup plus difficile à enlever qu’une tumeur friable, et peut être approchée par le T2 : une image hétérogène en hypersignal T2 sera friable, une image hypointense serait ferme ou sur la diffusion (un hyposignal serait en faveur d’une tumeur ferme) 4 même si cela reste encore discutée 70.
Particularité selon le type d’adénome
L’adénome somatotrope
Les adénomes somatotropes richement granulaires (densely granulated) répondent plus favorablement aux traitements médicaux. Ces adénomes pourraient être dépistés par un hyposignal en séquence T2. Cela nécessite néanmoins confirmation par des études complémentaires 58.
En l’absence d’adénome retrouvé sur l’IRM hypophysaire, un dosage de GH-RH avec scanner thoraco-abdominal, et un Octréoscan seront pratiqués pour dépister les tumeurs responsables d’une sécrétion ectopique.
L’adénome corticotrope
L’imagerie par IRM hypophysaire est de règle dans les syndromes ACTH dépendant (figure 5).
Des clichés dynamiques et éventuellement une séquence écho de gradient 3D pondérée en T1 (ED3D) peuvent être utiles. L’adénome est souvent un microadénome de quelques millimètres encore intra-hypophysaire 55, rarement des macroadénomes (évolution d’un adénome corticotrope silencieux) 63. L’absence d’image intrasellaire doit faire rechercher un adénome ectopique (adénome de la tige, parasellaire, sphénoïdal ou pharyngé) 34,47.
Si l’IRM est considérée comme normale, la réalisation d’un cathétérisme des sinus pétreux inférieurs (SPI) doit être discutée (figure 5). L’indication de cet examen doit néanmoins être bien évaluée, car il s’agit d’un cathétérisme chez des patients au terrain vasculaire fragile (risque d’AVC). Après cathétérisme des sinus pétreux inférieurs (SPI), il est donc possible de doser l’ACTH au plus près de l’hypophyse et de définir un ratio ACTH dans les SPI ou valeur centrale sur la valeur périphérique qui supérieur spontanément à 2 ou à 3 après stimulation au CRH ou à la Desmopressine est en faveur d’une origine hypophysaire 5,12. La valeur du test dans la latéralisation avec la recherche d’un gradient d’ACTH droit/gauche est par contre peu fiable 79.
Les adénomes hypophysaires atypiques ou carcinomes
Le bilan IRM hypophysaire doit être complété par un bilan d’extension an cas de suspicion d’adénome atypique. Une IRM crânio-spinale et un PET-scan seront demandés à la recherche de localisations secondaires qui seules assureront le diagnostic de carcinome hypophysaire.
L’apoplexie pituitaire
Il n’y a pas de particularité, l’IRM représentant l’imagerie de référence. Le scanner cérébral est toutefois souvent pratiqué en premier lieu dans un contexte d’urgence avec l’hypothèse d’une hémorragie méningée éventuelle. Il montrera une tumeur sellaire souvent en hyperdensité. L’IRM montrera une image de tumeur plus ou moins infarcie (hyposignal T1, hypersignal T2) et hémorragique (hypersignal T1 et T2 initialement) où l’on recherchera l’extension extrasellaire et un éventuel résidu adénomateux (figure 10). Un épaississement muqueux du sinus sphénoïdal est fréquemment noté parfois mis en relation avec la sévérité de l’apoplexie 2,42.
Diagnostics différentiels
Le patient est parfois adressé avec un faux diagnostic d’adénome hypophysaire qu’il faut redresser.
Ainsi, d’autres tumeurs sellaires ou suprasellaires peuvent simuler un adénome. Les plus fréquentes restent des tumeurs bénignes tels le méningiome ou le craniopharyngiome (figures 11).
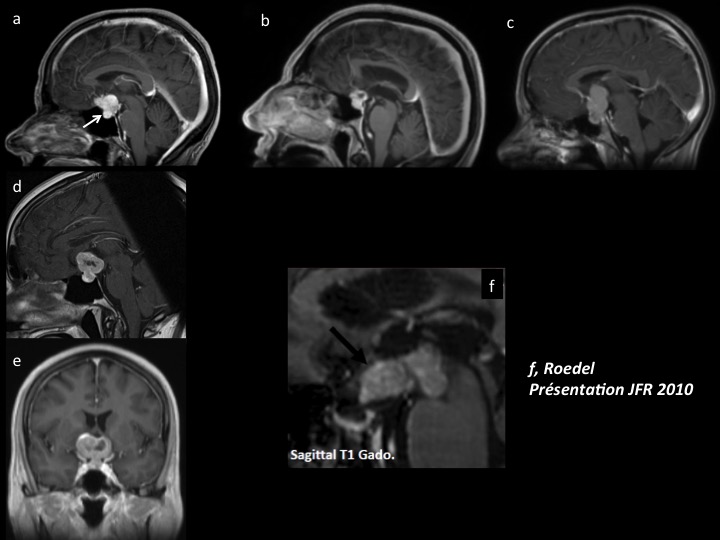
– Le méningiome du tubercule de la selle peut mimer un adénome. Il n’y a pas de critères cliniques spécifiques. La discordance radiologique entre une large partie suprasellaire et une petite selle turcique dans laquelle l’hypophyse reste visible redressera le diagnostic. Le signe de la queue de comète avec une prise de contraste large sur l’étage antérieur et moyen attirera aussi l’attention (figure 11a).
– Le craniopharyngiome : il s’agit d’un diagnostic de faible probabilité chez l’adulte (il existe néanmoins un pic au alentour de la 5ième décennie). L’insuffisance antéhypophysaire est plus fréquente mais surtout il peut exister une insuffisance post-hypophysaire totalement exceptionnelle en cas d’adénome hypophysaire. Le craniopharyngiome sellaire et suprasellaire dans sa forme purement kystique pourra être difficile à distinguer d’un adénome kystique (figure 11c). La forme classique mixte kystique-charnue avec quelques calcifications comme les formes suprasellaires sont par contre aisées à différencier d’un adénome (figure 11b).
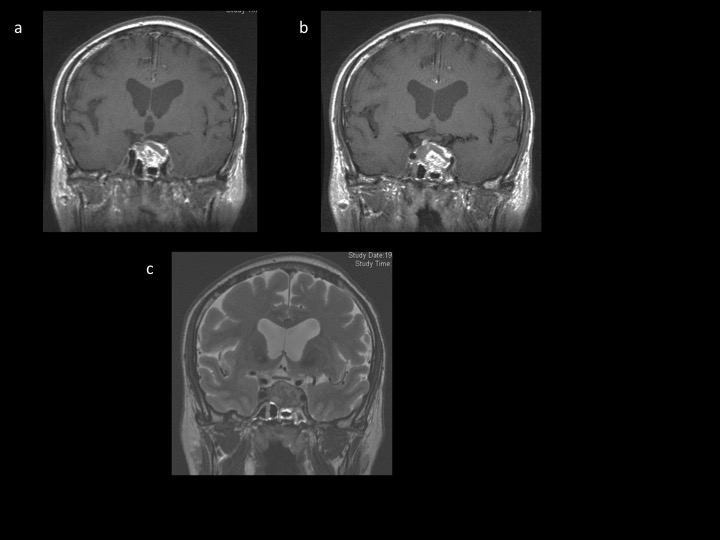
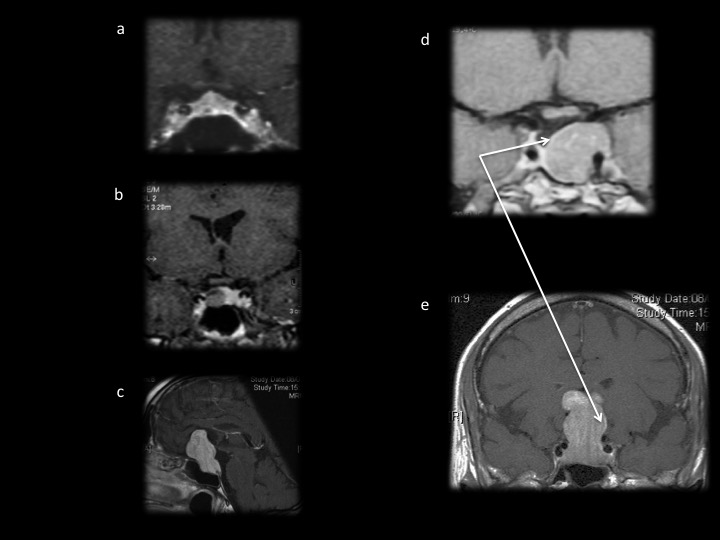
– Les autres types tumoraux sont rares. Les métastases de la région hypophysaire sont probablement sous-estimées (figures 11 d et e). Le contexte clinique (contexte oncologique, une insuffisance post-hypophysaire, paralysie oculomotrice) aidera à être méfiant et à évoquer le diagnostic car l’aspect radiologique n’est pas spécifique. Une lyse osseuse devra toutefois faire suspecter une étiologie maligne. Les aspects peuvent être variable entre un élargissement de la tige pituitaire ou tumeur sellaire ou suprasellaire. Le lymphome et le germinome atteignent surtout la région infundibulo-tubérienne avec une forte prise de contraste et souvent une atteinte anté ou posthypophysaire clinique (figure 11f). L’atteinte bipolaire est caractéristique des tumeurs germinales.
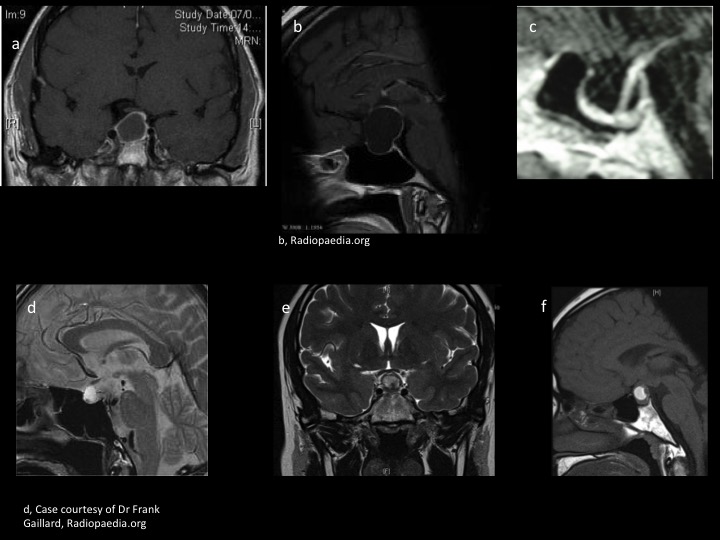
– La distinction entre adénomes kystiques (figure 12 a) et kystes de la région sellaire peut poser des problèmes dans la pratique (figure 12). Le kyste de la poche de Rathke est en hypersignal T1 dans la moitié des cas (contenu protéiforme), hypersignal T2 avec un nodule intrakystique ne prenant pas le produit de contraste surtout visible en T2 (figures 12 d, e, f). Le diagnostic de selle vide est posé devant une déhiscence du diaphragme sellaire avec visualisation de l’hypophyse plaquée au fond d’une selle turcique « ballonnisée ». Cela peut accompagner une hydrocéphalie chronique figure 12 c). Le kyste arachnoïdien sellaire et suprasellaire (figure 12 b) donnera cette même image d’hypophyse au fond de la selle mais avec un développement suprasellaire compressif. Il s’agit souvent de sujet d’âge moyen ou âgé. Le kyste épidermoïde ou dermoïde, diagnostic différentiel des kystes arachnoïdiens est souvent suprasellaire et la séquence de diffusion pourra aider au diagnostic (hyposignal et non hyper ne s’agissant pas de liquide).
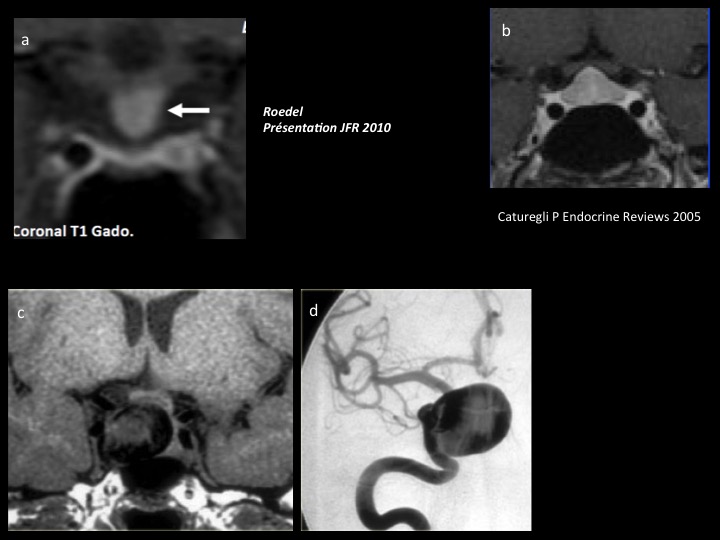
Les étiologies pseudotumorales inflammatoires (neurohypophysite, histiocytose, sarcoidose...) sont suprasellaires avec élargissement de la tige et fréquemment un diabète insipide dont nous avons dit qu’il devait faire remettre en question la diagnostic d’adénome (figures 13). Un diagnostic de pathologie inflammatoire peut être plus difficile, c’est l’adénohypophysite de la femme enceinte (fig. 13 b). Survenant dans le dernier trimestre ou dans le postpartum récent, l’image peut en imposer pour un adénome. Il faut y penser systématiquement dans le contexte.
L’abcès hypophysaire est rare et donnera une image en hypo signal T1 avec rehaussement périphérique dans un contexte hyperthermique et d’insuffisance hypophysaire.
Les exceptionnelles étiologies vasculaires doivent être présentes à l’esprit du fait des conséquences potentielles chirurgicales. Plus que l’anévrysme de la communicante antérieur plongeant, l’anévrysme carotido-caverneux déformant la selle peut être trompeur (fig 13 c et d).
++++
5. Traitements
5.1 Traitements médicaux
Le prolactinome
Le prolactinome reste une tumeur de prime abord de traitement médical.
Le principe est d’utiliser l’action inhibitrice de la Dopamine via les récepteurs dopaminergiques localisés sur la surface des cellules sécrétant la PRL. L’inhibition produit un défaut de sécrétion avec diminution des vésicules de stockage et donc diminution du volume tumoral. L’action est cytostatique.
Les agonistes dopaminergiques apparaissent à la fin des années 1970 avec le Parlodel® (Bromocriptine, prise quotidienne). La deuxième génération est représentée par le Norprolac® (Quinagolide, prise quotidienne) et la troisième depuis le début des années 2000 par le Dostinex® (Cabergoline, prise hebdomadaire ou quotidienne).
Le Dostinex® est actuellement le traitement de premier choix, sauf en cas de désir de grossesse, car il est plus efficace et mieux toléré que les autres agonistes de la dopamine. Les effets secondaires de ces agonistes sont : l’hypotension artérielle, des troubles digestifs avec nausées, vomissements, surtout à l’induction du traitement. Il est actuellement recommandé de réaliser une échocardiographie annuelle par le laboratoire en raison de la survenue d’atteinte valvulaire.
L’effet anti-sécrétoire et anti-tumoral est rapide, en quelques semaines, avec une chute de la PRL et une diminution de plus de 50% du volume tumoral (figure 7). Cependant, les traitements médicaux cytostatiques devront souvent poursuivi au moins jusqu’à la ménopause. Un sevrage peut être tenté après au moins 3 ans d’hypersécrétion et d’IRM normalisées. Vingt à trente pour cent des patientes ne récidiveraient pas 11,17. Les vraies résistances aux agonistes dopaminergiques sont rares entre 5 et 10%)78.
Particularités de la grossesse :
– Pour les microadénomes ou « petit » macroadénomes sans proximité avec le chiasma, la fertilité est restaurée dans 90% avec la baisse de l’hyperprolactinémie. En cas de projet de grossesse, un traitement par Parlodel sera préféré en raison de l’ancienneté de ce traitement, mais aucun effet tératogène n’a été relevé pour la cabergoline (Lebbe Clin Endo 2010) 41. Le risque d’augmentation de volume est très faible (<2%) et le traitement est arrêté pendant la grossesse sans surveillance particulière (pas de dosage de la PRL en raison de l’augmentation physiologique gravidique), l’allaitement sera permis. Un bilan biologique et une IRM seront effectués au décours.
– Pour les macroadénomes, le risque d’augmentation de volume justifie la poursuite du traitement médical pendant la grossesse. La surveillance sera visuelle avec un examen ophtalmologique tous les trimestres et une IRM en cas de signes compressifs. L’allaitement sera contre-indiqué.
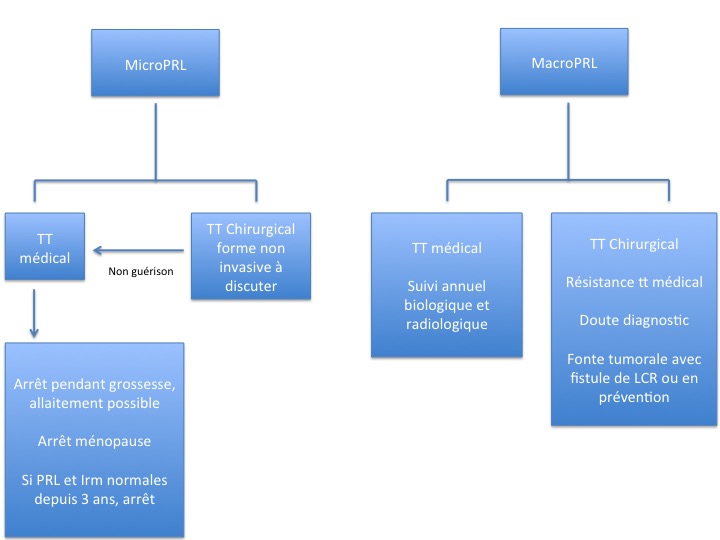
L’algorithme de la figure 14 résume la prise en charge actuelle des prolactinomes.
L’adénome somatotrope
Les échecs de la chirurgie ou les adénomes de volume moyen ou petit avec envahissement de la loge caverneuse seront traités médicalement (algorithme de prise en charge, figure 4).
– Les traitements de première ligne sont : les analogues de la somatostatine (AS) de première génération (Octréotide, Lanréotide) agissant sur les récepteurs SST2 présents sur la membrane de la cellule tumorale dans 90% des adénomes somatotropes)16. Ces analogues suppriment la production de GH diminuant ainsi le volume cellulaire et parfois le volume tumoral. L’action est néanmoins cytostatique et par définition les analogues ne guérissent pas les patients. Ce traitement nécessite une injection IM ou sous-cutanée profonde toutes les 4 semaines. Un premier bilan (GH-IGF-1 +/- IRM) sera réalisé à la veille de la 4ème injection afin d’évaluer la sensibilité au traitement et permettre son adaptation. Une augmentation des doses et/ou une augmentation de la fréquence des injections seront discutées en l’absence de contrôle de l’hypersécrétion. Les effets secondaires des traitements sont principalement digestifs, par diminution de la sécrétion d’enzymes pancréatiques avec malabsorption et diarrhées ainsi que les lithiases vésiculaires. La prescription d’enzymes pancréatiques encadrant les injections d’AS permet de soulager les patients et ces effets digestifs s’amenuisent avec le temps. Il convient aussi de prendre en compte les coûts élevés de ces traitements.
– L’efficacité est corrélée au volume et à l’hypersécrétion avec en moyenne 50% de répondeurs à un an et 30% de réduction de volume tumoral après ce délai. Bien que l’effet thérapeutique se poursuive avec le temps 44, une réponse insuffisante après 6 mois de traitement doit faire poser la question d’un traitement complémentaire ou d’un changement de stratégie thérapeutique.
Les critères de contrôle sont identiques aux critères de guérison (GH moyenne <2,5 μg/l (7,5 mI/l), IGF1 normale pour l’âge, Nadir GH/HGPO : <0,4μg/l (1,2 mI/l)).
– Les traitements de deuxième ligne sont :
- les agonistes dopaminergiques : ces thérapeutiques sont d’utilisation évidente en cas d’adénome mixte GH-PRL, mais peuvent être utiles, pour traiter des adénomes à GH purs en raison de la présence possible de récepteurs dopaminergiques membranaires. Ils sont utilisés en association avec les AS en cas de maladie non contrôlée, avec élévation modérée de l’IGF1 (<1,3 fois la normale) mais peuvent également être utilisés en première ligne en cas d’acromégalie modérée.
- Le nouvel AS, pasiréotide®, actif sur 4 des 5 récepteurs de la SST est une option de deuxième ligne et les résultats préliminaires montrent qu’il pourrait être plus efficace dans le contrôle de l’hypersécrétion et du volume tumoral et également en cas de résistance aux AS classiques 56.
- En cas d’échec des AS, l’antagoniste de la GH (fixation et blocage des récepteurs à GH hépatique) ou PEGVISOMANT® permet le contrôle d’au moins 70% des patients mais n’exerce aucun effet anti-tumoral. Il est utilisé seul en cas de résistance aux AS ou en association avec les AS en cas de volume tumoral important ou de signes tumoraux gênants type céphalées à raison d’une injection journalière. La surveillance se fait sur les taux d’IGF-1 uniquement avec des bilans hépatiques réguliers (en début de traitement, à 15 jours et tous les 6 mois) et une IRM à 6 mois. Des augmentations de volume ont été décrites à l’initiation des traitements ou à l’arrêt des AS.
L’adénome corticotrope
Sur le plan thérapeutique, l’objectif est de supprimer l’hypersécrétion corticotrope, fusse au prix d’un déficit antéhypophysaire et de traiter les complications.
En cas d’échec de la chirurgie, un traitement médical sera mis en œuvre avec au choix sans consensus les analogues de la somatostatine d’action hypophysaire : pasiréotide® (effets secondaires communs aux analogues de la SMS avec risque de décompensation diabétique) ou les anticortisoliques d’action surrénalienne : OP’-DDD, la métopirone ou le kétonazole avec risque d’hépatite pour les antifongiques. Ce traitement sera mis dans l’attente d’une action thérapeutique pérenne qui sera discutée au cas par cas en RCP hypophysaire : reprise chirurgicale hypophysaire, surrénalectomie bilatérale (parfois associée à une radiothérapie hypophysaire) ou une radiothérapie hypophysaire seule surtout en cas de cible individualisée. Dans ce cas, le traitement médical sera maintenu le temps d’obtention de l’effet de la radiothérapie qui se manifeste entre 12 et 18 mois.
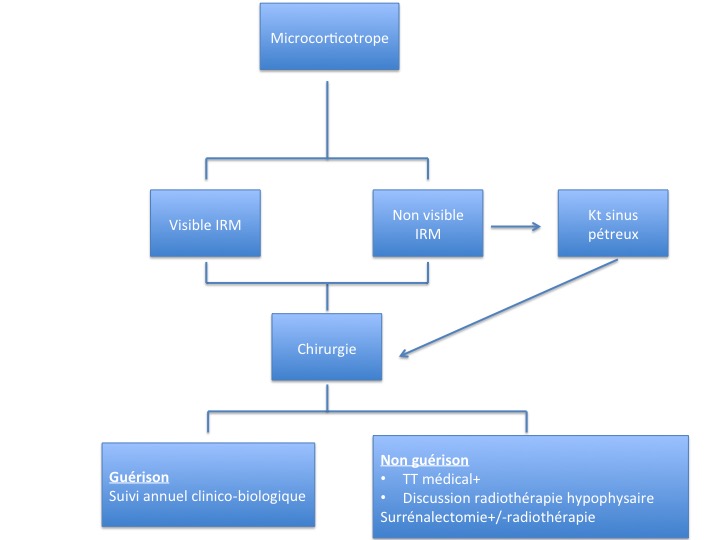
L’algorithme de la figure 15 résume la stratégie de la prise en charge des microcorticotropes.
L’adénome thyréotrope
Le traitement peut être chirurgical ou médical par analogues de la somatostatine et vise à restaurer l’euthyroidisme.
Les séries rapportées de la littérature concernent un petit nombre de patients et aucune conclusion définitive ne peut être donnée.
Les AS sont efficaces dans le contrôle de l’hypersécrétion avec 40% de diminution de volume tumoral car tous les adénomes thyréotropes expriment fortement les récepteurs de sous-type 2 et 5 16. Ces AS se discutent en première option lors d’adénomes de volume moyen sans troubles visuels d’autant qu’il existe un envahissement latéral.
Les adénomes hypophysaires non sécrétants ou non fonctionnels (gonadotropes et « silencieux »)
Aucun traitement médical n’a démontré d’efficacité pérenne malgré quelques articles rapportant quelques résultats avec les agonistes dopaminergiques ou les analogues de la somatostatine 18,19.
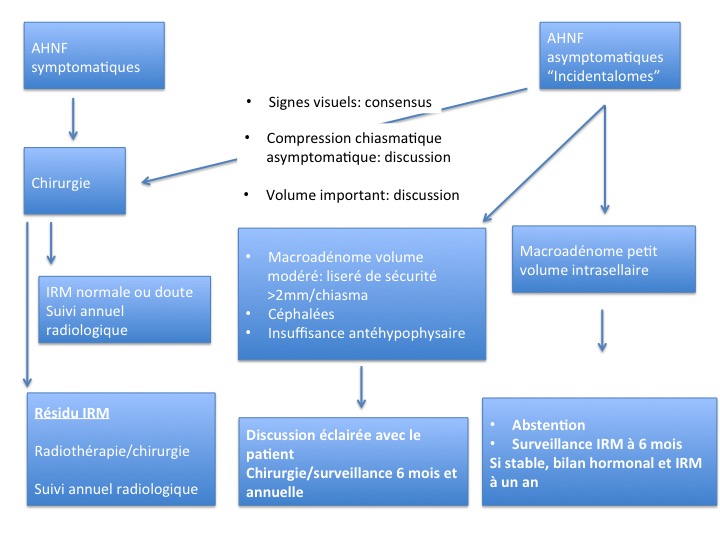
La stratégie est donc surtout chirurgicale et résumée par l’algorithme de la figure 16.
Les adénomes atypiques ou carcinomes hypophysaires
Les données sont encore plus rares concernant le Bévacizumab (anti-angiogénique anti-VEGF) (ortiz pituitary 2012) et des anti-mTOR 36,54. Certes, il existe une base physiopathologique avec hyperexpression de facteurs angiogéniques et une activation de la voie PI3K/AKT/m-TOR dans certains adénomes agressifs, mais très peu de patients ont été traités à ce jour 33.
L’apoplexie pituitaire
Le traitement reste sujet à controverse et a fait l’objet d’une conférence de consensus par la société anglaise d’endocrinologie60. Un transfert dans centre de référence hypophysaire est la règle. La controverse porte sur la question « indication chirurgicale ou traitement conservateur » car la littérature est confuse sans étude randomisée. Schématiquement, seront considérés pour un traitement conservateur les patients sans troubles visuels sévères ou en aggravation ni troubles de la vigilance. Les paralysies oculomotrices ne sont pas des indications chirurgicales en l’absence de problème d’AV ou de CV.
++++
5.2 Chirurgie
La technique fait l’objet d’un chapitre spécifique auquel le lecteur peut se rapporter.
Le prolactinome
En raison du caractère cytostatique des traitements médicaux, dans les cas de microadénomes ou petit macroadénomes enclos, la chirurgie peut être proposée comme alternative thérapeutique car la chirurgie pourra être curatrice évitant d’éventuels effets secondaires ou l’incertitude d’un traitement de longue durée. Dans ces formes spécifiques, le taux de guérison avoisine les 90% pour un risque opératoire notamment d’insuffisance antéhypophysaire faible (inférieur à 5%) 71. Les rares cas de résistance au traitement médical (5 à 10%) 78 et les microadénomes GH-PRL sont aussi des indications chirurgicales. La chirurgie des micro-prolactinomes est délicate, nécessitant une expérience chirurgicale hypophysaire importante, à l’instar de la maladie de Cushing.
Pour les autres macroadénomes, la chirurgie a comme indications la résistance au traitement médical observée dans moins de 10% des cas (la réduction tumorale restaurera la sensibilité aux agonistes de la dopamine dans la grande majorité des cas, 59 et les apoplexies pituitaires (ramollissement hémorragique intratumoral) avec troubles visuels. Parfois, la tumeur a détruit les enveloppes ostéodurales et la « fonte » tumorale peut être à l’origine d’une fistule de LCS. Une chirurgie de réparation ou mieux préventive peut se discuter, mais dans les faits, les indications sont rares. Dans les cas de tumeurs invasives, la gestion de ce risque de fistule peut être faite en utilisant une faible posologie permettant de conserver un résidu tumoral non compressif, obturant la selle turcique.
L’adénome somatotrope
Sur le plan thérapeutique, en raison de la morbidité et de la mortalité associées à l’hypersécrétion de GH, toute acromégalie doit d’être traitée (l’algorithme de prise en charge est résumé dans la figure 4).
La chirurgie se discute en première intention, comme seul traitement capable de guérir le patient rapidement et la décision fonction de l’envahissement des loges caverneuses. En l’absence d’envahissement de celle-ci, la chirurgie représente le « gold-standard ». S’il existe un doute sur l’invasion, une chirurgie sera proposée au patient en expliquant le risque d’échec. Elle est la règle lors de troubles visuels même en cas d’envahissement des loges caverneuses car les traitements médicaux sont longs à agir et la réduction du volume tumoral est moins importante que celle des prolactinomes sous agoniste de la dopamine. En cas d’invasion évidente, l’attitude dépendra du volume tumoral. En effet, si le volume est important (2 à 3 cm), l’hypersécrétion de GH sera difficile à contrôler rapidement avec le seul traitement médical ; dans ces conditions une réduction volumétrique première sera discutée, car la réduction tumorale préopératoire permet de réduire les délais de contrôle de la maladie.
Pour le chirurgien, une attention particulière sera portée sur le trajet des carotides (dans leur portion C5 paraclinoïdienne) ayant tendance à revenir sur la ligne médiane (« Kissing arteries ») entrainant un risque vasculaire supplémentaire 27. L’anatomie nasale est aussi particulière avec des structures hypertrophiées et rigides. Une résection du cornet moyen peut être nécessaire pour « passer », améliorant aussi la perméabilité de la fosse nasale et le confort respiratoire des acromégales.
Les résultats de la chirurgie sont inversement corrélés aux taux pré-opératoires de GH, au volume tumoral, à l’envahissement des loges caverneuses et à l’expérience du chirurgien 7. Les taux de guérison postopératoire sont de l’ordre de 80% pour les micro ou macroadénomes enclos, mais chutent à 40-50% pour des macroadénomes intra et extrasellaires 10.
Le traitement médical préopératoire peut améliorer les conditions anesthésiologiques (difficultés d’intubation, insuffisance cardiaque), mais il persiste une controverse quant à son utilité pour optimiser le taux de guérison chirurgical 57.
L’adénome corticotrope
Sur le plan thérapeutique, l’objectif est de supprimer l’hypersécrétion corticotrope, fusse au prix d’un déficit antéhypophysaire et de traiter les complications.
La chirurgie est le traitement de première intention. Elle doit être réalisée par un neurochirurgien spécialisé en chirurgie hypophysaire qui réalisera une adénomectomie élargie ou une hémi-hypophysectomie. Pour ce faire, il enlèvera l’adénome et une zone d’antéhypophyse juxtatumorale pour s’assurer d’une exérèse complète. Il s’agit de la chirurgie hypophysaire qui requiert le plus d’expérience. Dans presque un tiers des cas, le chirurgien opérera l’hypophyse sans image visible sur l’IRM pré opératoire 68. Il pourra être aidé dans sa recherche de la tumeur par un examen extemporané réalisé par un pathologiste hypophysaire expérimenté.
Le meilleur critère de guérison postopératoire est l’inertie corticotrope qui peut se prolonger plusieurs mois voir une année après la chirurgie. En effet, la tumeur enlevée, les cellules antéhypophysaires normales qui ont été freinées par l’hypersécrétion de cortisol resteront quiescentes pendant un laps de temps non prévisible. L’éducation thérapeutique s’avère donc essentielle, le patient étant désormais en insuffisance corticotrope. Le taux de guérison après chirurgie trans-sphénoïdale est d’environ 75-80% dans les microadénomes 68.
Cette phase postopératoire est difficile pour tous les patients ayant eu l’habitude de vivre avec un excès de cortisol ; le retour à un taux normal s’accompagne d’une grande asthénie, voire d’une dépression. Il convient d’en prévenir les patients, d’établir un soutien psychologique et de ne pas recourir à l’augmentation des doses d’hydrocortisone.
L’adénome thyréotrope
Il persiste une controverse mais schématiquement les formes intrasellaires encloses sans envahissement des loges caverneuses ou avec des troubles visuels sont chirurgicales et les autres indications sont discutables 76.
Les adénomes hypophysaires non sécrétants ou non fonctionnels (gonadotropes et « silencieux »)
Sur le plan thérapeutique , il n’y a pas de traitement médical efficace. En cas de troubles visuels, l’indication chirurgicale est indiscutable avec 90% d’amélioration ophtalmologique sauf atrophie compatibles avec une vie personnelle et professionnelle normale 50. L’insuffisance hypophysaire partielle récupère dans environ 25% des cas avec une morbidité d’environ 15% (13% de déficit antéhypophysaire, moins de 10% de diabète insipide) 50. En cas de déficits endocriniens sans troubles visuels ou d’incidentalome (découverte fortuite), l’indication chirurgicale est discutée : Si l’adénome menace les structures optochiasmatiques, une option chirurgicale est proposée aux patients et s’il n’existe pas de menace, un contrôle IRM à 6 mois puis annuelle est préconisée pour dépister une évolutivité, avec parallèlement surveillance ophtalmologique. La décision est partagée avec le patient, sachant que les résultats sont meilleurs si l’on intervient tôt avec dans notre expérience une absence de complications visuelles chez les patients indemnes en pré opératoire 51.
En cas de résidu (20 à 30% de résidus tumoraux postopératoires 50 ou de récidive postopératoire, la décision de traitement complémentaire discuté en RCP spécialisée en pathologie hypophysaire repose sur :
– Des critères anatomiques : volume et localisation du résidu. Une menace persistante sur les voies optiques, un volume important ou l’espoir d’une chirurgie complète pourra faire discuter une réintervention par voie basse ou haute.
– Des critères anatomopathologiques. Si l’adénome est « atypique » ou grade 2b selon les récentes classifications, un traitement complémentaire sera discuté, surtout si le patient est jeune
– De l’âge et du contexte clinique. Il est certain que chez le sujet jeune où l’objectif est le contrôle du résidu sur des décennies, la tendance sera d’être plus « agressif » que chez le sujet âgé. Néanmoins il convient sans doute de ne pas se « précipiter » vers une décision de radiothérapie, notamment dans les cas de résidus intra-caverneux. En effet nombre de résidus restent stables ou n’augmentent que 4 à 5 ans plus tard.
Les adénomes atypiques ou carcinomes hypophysaires
Les indications d’interventions itératives que cela soit sur le site primaire ou sur les métastases se discutent au cas par cas en RCP multidisciplinaire.
L’apoplexie pituitaire
Le traitement reste sujet à controverse et a fait l’objet d’une conférence de consensus par la société anglaise d’endocrinologie 60. Un transfert dans centre de référence hypophysaire est la règle. La controverse porte sur la question « indication chirurgicale ou traitement conservateur » car la littérature est confuse sans étude randomisée. Schématiquement, sera considéré pour la chirurgie par voie trans-sphénoïdale : en urgence, un patient ayant des troubles visuels sévères (AV, CV) ou des troubles de la vigilance (compression hypothalamique ou hydrocéphalie) mais l’intervention peut être différée de quelques heures car elle doit être faite par un neurochirurgien référent. En semi-urgence, le patient qui a des troubles importants (AV, CV), s’aggravant ou restant stables après un recul de quelques jours (maximum 8 jours). La persistance de céphalées importantes et résistantes aux traitements médicaux peut faire discuter d’une chirurgie donc l’action antalgique est spectaculaire. Les paralysies oculomotrices ne sont pas des indications en l’absence de problème d’AV ou de CV.
++++
5.3 La radiothérapie
Le prolactinome
La radiothérapie n’est utilisée que dans les rares cas de résistance au traitement médical avec des taux de contrôle semblant intéressant (entre 62,5 et 100%) Toutefois, les cohortes sont petites pour établir une conclusion définitive et l’efficacité de la chirurgie et surtout des traitements médicaux rendent le recours à la radiothérapie très rare 64,69,72.
L’adénome somatotrope
La radiothérapie est discutée chez le sujet jeune ou d’âge moyen pour à terme pouvoir guérir les patients. Son délai d’action est long, en moyenne 25% de patients sont contrôlés à 5 ans et 50% à 10 ans 52. Aucune technique n’a montré une efficacité supérieure à l’autre (radiothérapie fractionnée stéréotaxique ou conventionnelle et radiochirurgie) 14. Le choix de la technique est donc fonction de la disponibilité des machines et de la cible à traiter (radiochirurgie en cas de petite tumeur, ou de reliquat tumoral localisée ?). Le taux d’insuffisance antéhypophysaire post-radique, d’environ 25% à 5 ans, justifie un suivi annuel.
L’adénome corticotrope
Indiquée en cas d’échec du traitement chirurgical, elle portera soit sur l’hypophyse surtout si une cible est individualisée soit complétera une surrénalectomie en prévention d’un syndrome de Nelson. Comme pour les autres sous-types d’adénomes, la technique (radiothérapie conformationnelle fractionnée, stéréotaxique fractionnée ou radiochirurgie) dépendra du volume, de la distance vis à vis des voies visuelles, de l’expérience de l’équipe sans prédominance indiscutable d’une technique l’une par rapport à une autre. Lest taux de contrôle pour la radiothérapie conventionnelle sont de 83% à 4 ans 28et varie entre 17 à 80% pour les séries de radiochirurgie en raison de critères de guérison diverses 13,40. L’effet est retardé dans le temps avec une moyenne de 2 ans avant d’obtenir un contrôle de l’hypersécrétion.
L’adénome thyréotrope
La radiothérapie semble décevante, mais la encore le nombre de cas traités est faible 76.
Les adénomes hypophysaires non sécrétants ou non fonctionnels (gonadotropes et « silencieux »)
En cas de résidu (20 à 30% de résidus tumoraux post-opératoires 50 ou de récidive postopératoire, la décision de traitement complémentaire discuté en RCP spécialisée Hypophyse repose sur :
– Des critères anatomiques : volume et localisation du résidu. Une menace persistante sur les voies optiques, un volume important ou l’espoir d’une chirurgie complète pourra faire discuter une ré-intervention par voie basse ou haute.
– Des critères anatomopathologiques. Si l’adénome est « atypique » ou grade 2b selon les récentes classifications, un traitement complémentaire sera discuté, surtout si le patient est jeune
– De l’âge et du contexte clinique. Il est certain que chez le sujet jeune où l’objectif est le contrôle du résidu sur des décennies, la tendance sera d’être plus « agressif » que chez le sujet âgé. Néanmoins il convient sans doute de ne pas se « précipiter » vers une décision de radiothérapie, notamment dans les cas de résidus intra-caverneux.
Aucun type de radiothérapie (radiothérapie multifractionnée conventionnelle, radiochirurgie, radiothérapie stéréotaxique fractionnée) n’a démontré une efficacité supérieure à un autre. Plus que l’outil technique, c’est l’expérience de l’équipe qui prime. La « vraie » radiochirurgie en dose unique (les études les plus nombreuses concernent le gammaknife) est réservée aux petites cibles à distance des voies visuelles (au moins 3 mm). En effet les risques visuels et endocriniens post-radiques sont corrélés au volume tumoral irradié. Il en est de même des risques d’échappement tumoral à long terme 15,66. Globalement, le taux de contrôle tumoral est d’environ 90% à 5 ans mais peut chuter à 80% à 10 ans lorsque les résidus tumoraux sont plus volumineux > 4 ml. Sur de plus gros volume ou selon la disponibilité des techniques, une radiothérapie stéréotaxique hypofractionnée 31 ou en fractionnement classique seront choisis avec un taux de contrôle similaires 65. Le risque d’insuffisance antéhypophysaire est d’environ 25% entre 3 et 5 ans. Il est à noter que les échappements peuvent se voir à long et très long terme ce qui valide une attitude de surveillance de longue durée au delà des 10 ans.
6. Suivi
Si l’on tient compte des récidives à moyen ou long terme des hypersécrétions (12% des prolactinomes traités chirurgicalement par exemple) ou des résidus tumoraux des adénomes non fonctionnels (évolution à 5 voire 10 ans et au delà y compris après radiothérapie), la surveillance doit être sur le long terme. Un rythme annuel avec une consultation, un bilan biologique et souvent une IRM est souvent adopté par les différentes équipes.
++++
7. Bibliographie
1. Agrawal NK, Goyal R, Rastogi A, Naik D, Singh SK : Thyroid hormone resistance. Postgrad Med J 84:473–477, 2008 C4
2. Arita K, Kurisu K, Tominaga A, Sugiyama K, Ikawa F, Yoshioka H, et al. : Thickening of sphenoid sinus mucosa during the acute stage of pituitary apoplexy. J Neurosurg 95:897–901, 2001 C4
3. Ayuk J, McGregor EJ, Mitchell RD, Gittoes NJL : Acute management of pituitary apoplexy—surgery or conservative management ? Clin Endocrinol (Oxf) 61:747–752, 2004 C4
4. Bahuleyan B, Raghuram L, Rajshekhar V, Chacko AG : To assess the ability of MRI to predict consistency of pituitary macroadenomas. Br J Neurosurg 20:324–326, 2006 B2
5. Bessac L, Bachelot I, Vasdev A, Martinie M, Bonnier L, Chabre O, et al. : [Catheterization of the inferior petrosal sinus. Its role in the diagnosis of Cushing’s syndrome. Experience with 23 explorations]. Ann Endocrinol (Paris) 53:16–27, 1992 C4
6. Biller BMK, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertherat J, et al. : Treatment of adrenocorticotropin-dependent Cushing’s syndrome : a consensus statement. J Clin Endocrinol Metab 93:2454–2462, 2008 A1
7. Bourdelot A, Coste J, Hazebroucq V, Gaillard S, Cazabat L, Bertagna X, et al. : Clinical, hormonal and magnetic resonance imaging (MRI) predictors of transsphenoidal surgery outcome in acromegaly. Eur J Endocrinol 150:763–771, 2004 B2
8. Brucker-Davis F, Oldfield EH, Skarulis MC, Doppman JL, Weintraub BD : Thyrotropin-secreting pituitary tumors : diagnostic criteria, thyroid hormone sensitivity, and treatment outcome in 25 patients followed at the National Institutes of Health. J Clin Endocrinol Metab 84:476–486, 1999 C4
9. Brue T, Delemer B : Diagnosis and management of hyperprolactinemia : expert consensus - French Society of Endocrinology. Ann Endocrinol (Paris) 68:58–64, 2007 A1
10. Campbell PG, Kenning E, Andrews DW, Yadla S, Rosen M, Evans JJ : Outcomes after a purely endoscopic transsphenoidal resection of growth hormone-secreting pituitary adenomas. Neurosurg Focus 29:E5, 2010 C4
11. Casanueva FF, Molitch ME, Schlechte JA, Abs R, Bonert V, Bronstein MD, et al. : Guidelines of the Pituitary Society for the diagnosis and management of prolactinomas. Clin Endocrinol (Oxf) 65:265–273, 2006 A1
12. Castinetti F, Morange I, Dufour H, Jaquet P, Conte-Devolx B, Girard N, et al. : Desmopressin test during petrosal sinus sampling : a valuable tool to discriminate pituitary or ectopic ACTH-dependent Cushing’s syndrome. Eur J Endocrinol 157:271–277, 2007 C2
13. Castinetti F, Nagai M, Dufour H, Kuhn J-M, Morange I, Jaquet P, et al. : Gamma knife radiosurgery is a successful adjunctive treatment in Cushing’s disease. Eur J Endocrinol 156:91–98, 2007 C2
14. Chanson P, Bertherat J, Beckers A, Bihan H, Brue T, Caron P, et al. : French consensus on the management of acromegaly. Ann Endocrinol (Paris) 70:92–106, 2009 A1
15. Chen Y, Li ZF, Zhang FX, Li JX, Cai L, Zhuge QC, et al. : Gamma knife surgery for patients with volumetric classification of nonfunctioning pituitary adenomas : a systematic review and meta-analysis. Eur J Endocrinol 169:487–495, 2013 B2
16. Chinezu L, Vasiljevic A, Jouanneau E, François P, Borda A, Trouillas J, et al. : Expression of somatostatin receptors, SSTR2A and SSTR5, in 108 endocrine pituitary tumors using immunohistochemical detection with new specific monoclonal antibodies. Hum Pathol 45:71–77, 2014 B2
17. Colao A, Di Sarno A, Guerra E, Pivonello R, Cappabianca P, Caranci F, et al. : Predictors of remission of hyperprolactinaemia after long-term withdrawal of cabergoline therapy. Clin Endocrinol (Oxf) 67:426–433, 2007 B2
18. Colao A, Di Somma C, Pivonello R, Faggiano A, Lombardi G, Savastano S : Medical therapy for clinically non-functioning pituitary adenomas. Endocr Relat Cancer 15:905–915, 2008 C4
19. Colao A, Filippella M, Di Somma C, Manzi S, Rota F, Pivonello R, et al. : Somatostatin analogs in treatment of non-growth hormone-secreting pituitary adenomas. Endocrine 20:279–283, 2003 C4
20. Cottier JP, Destrieux C, Brunereau L, Bertrand P, Moreau L, Jan M, et al. : Cavernous sinus invasion by pituitary adenoma : MR imaging. Radiology 215:463–469, 2000 B2
21. Cushing H : The pituitary and ist disorders. Classics of Medecine., Birmingham, 1979 C4
22. Daneshdoost L, Gennarelli TA, Bashey HM, Savino PJ, Sergott RC, Bosley TM, et al. : Identification of gonadotroph adenomas in men with clinically nonfunctioning adenomas by the luteinizing hormone beta subunit response to thyrotropin-releasing hormone. J Clin Endocrinol Metab 77:1352–1355, 1993 C4
23. Daneshdoost L, Gennarelli TA, Bashey HM, Savino PJ, Sergott RC, Bosley TM, et al. : Recognition of gonadotroph adenomas in women. N Engl J Med 324:589–594, 1991C4
24. DeLellis R, Lloyd R, Heitz P, Eng C : Tumours of endocrine organs, in World Health Organization classification of tumors, IARC. Lyon A1
25. Delgrange E, Trouillas J, Maiter D, Donckier J, Tourniaire J : Sex-related difference in the growth of prolactinomas : a clinical and proliferation marker study. J Clin Endocrinol Metab 82:2102–2107, 1997 B2
26. Dudziak K, Honegger J, Bornemann A, Horger M, Müssig K : Pituitary carcinoma with malignant growth from first presentation and fulminant clinical course—case report and review of the literature. J Clin Endocrinol Metab 96:2665–2669, 2011 C4
27. Ebner FH, Kuerschner V, Dietz K, Bueltmann E, Naegele T, Honegger J : Reduced intercarotid artery distance in acromegaly : pathophysiologic considerations and implications for transsphenoidal surgery. Surg Neurol 72:456–460 ; discussion 460, 2009 C4
28. Estrada J, Boronat M, Mielgo M, Magallón R, Millan I, Díez S, et al. : The long-term outcome of pituitary irradiation after unsuccessful transsphenoidal surgery in Cushing’s disease. N Engl J Med 336:172–177, 1997 C4
29. Galen : Galen on the usefulness of the parts of the body (Peri Chreias Moion), in Cornell Universiy Press. Ithaca, NY, 1968 C4
30. Garcia T, Sanchez S, Litré CF, Radoi C, Delemer B, Rousseaux P, et al. : Prognostic value of retinal nerve fiber layer thickness for postoperative peripheral visual field recovery in optic chiasm compression. J Neurosurg 121:165–169, 2014 C4
31. Iwata H, Sato K, Tatewaki K, Yokota N, Inoue M, Baba Y, et al. : Hypofractionated stereotactic radiotherapy with CyberKnife for nonfunctioning pituitary adenoma : high local control with low toxicity. Neuro-oncology 13:916–922, 2011B2
32. Jacob M, Raverot G, Jouanneau E, Borson-Chazot F, Perrin G, Rabilloud M, et al. : Predicting visual outcome after treatment of pituitary adenomas with optical coherence tomography. Am J Ophthalmol 147:64–70.e2, 2009 C4
33. Jia W, Sanders AJ, Jia G, Liu X, Lu R, Jiang WG : Expression of the mTOR Pathway Regulators in Human Pituitary Adenomas Indicates the Clinical Course. Anticancer Res 33:3123–3131, 2013 C4
34. Jouanneau E, Perrin G, Trouillas J : [Corticotroph microadenomas of the pituitary stalk. Diagnostic and therapeutic difficulties]. Neurochirurgie 48:215–222, 2002 C4
35. Jouanneau E : Les adénomes hypophysaires non fonctionnels : résultats tumoraux, endocriniens et ophtalmologiques à long terme. Thèse de Médecine, Université Lyon I, Lyon1999 C4
36. Jouanneau E, Wierinckx A, Ducray F, Favrel V, Borson-Chazot F, Honnorat J, et al. : New targeted therapies in pituitary carcinoma resistant to temozolomide. Pituitary 15:37–43, 2012 C4
37. Karavitaki N : Prevalence and incidence of pituitary adenomas. Ann Endocrinol (Paris) 73:79–80, 2012 C4
38. Knosp E, Steiner E, Kitz K, Matula C : Pituitary adenomas with invasion of the cavernous sinus space : a magnetic resonance imaging classification compared with surgical findings. Neurosurgery 33:610–617 ; discussion 617–618, 1993 C4
39. Lafferty AR, Chrousos GP : Pituitary tumors in children and adolescents. J Clin Endocrinol Metab 84:4317–4323, 1999 C4
40. Laws ER, Sheehan JP, Sheehan JM, Jagnathan J, Jane JA, Oskouian R : Stereotactic radiosurgery for pituitary adenomas : a review of the literature. J Neurooncol 69:257–272, 2004 C4
41. Lebbe M, Hubinont C, Bernard P, Maiter D : Outcome of 100 pregnancies initiated under treatment with cabergoline in hyperprolactinaemic women. Clin Endocrinol (Oxf) 73:236–242, 2010 C4
42. Liu JK, Couldwell WT : Pituitary apoplexy in the magnetic resonance imaging era : clinical significance of sphenoid sinus mucosal thickening. J Neurosurg 104:892–898, 2006 C4
43. Maartens NF : The history of the treatment of pituitary adenomas. Endocrine 28:9–26, 2005 C4
44. Maiza JC, Vezzosi D, Matta M, Donadille F, Loubes-Lacroix F, Cournot M, et al. : Long-term (up to 18 years) effects on GH/IGF-1 hypersecretion and tumour size of primary somatostatin analogue (SSTa) therapy in patients with GH-secreting pituitary adenoma responsive to SSTa. Clin Endocrinol (Oxf) 67:282–289, 2007 B2
45. Mancini T, Casanueva FF, Giustina A : Hyperprolactinemia and prolactinomas. Endocrinol Metab Clin North Am 37:67–99, viii, 2008 C4
46. Marie P : Sur deux cas d’acromégalie : hypertrophie singulière, non congénitale, des extrémités supérieures, inférieures et céphalique. 6:297–333, 1886 C4
47. Mason RB, Nieman LK, Doppman JL, Oldfield EH : Selective excision of adenomas originating in or extending into the pituitary stalk with preservation of pituitary function. J Neurosurg 87:343–351, 1997 C4
48. Melmed S, Colao A, Barkan A, Molitch M, Grossman AB, Kleinberg D, et al. : Guidelines for acromegaly management : an update. J Clin Endocrinol Metab 94:1509–1517, 2009 A1
49. Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, et al. : Diagnosis and treatment of hyperprolactinemia : an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 96:273–288, 2011 A1
50. Messerer M, De Battista JC, Raverot G, Kassis S, Dubourg J, Lapras V, et al. : Evidence of improved surgical outcome following endoscopy for nonfunctioning pituitary adenoma removal. Neurosurg Focus 30:E11, 2011 C4
51. Messerer M, Dubourg J, Raverot G, Bervini D, Berhouma M, George I, et al. : Non-functioning pituitary macro-incidentalomas benefit from early surgery before becoming symptomatic. Clin Neurol Neurosurg 115:2514–2520, 2013 C4
52. Minniti G, Scaringi C, Enrici RM : Radiation techniques for acromegaly. Radiat Oncol 6:167, 2011 C4
53. Ónnestam L, Berinder K, Burman P, Dahlqvist P, Engström BE, Wahlberg J, et al. : National incidence and prevalence of TSH-secreting pituitary adenomas in Sweden. J Clin Endocrinol Metab 98:626–635, 2013 C4
54. Ortiz LD, Syro LV, Scheithauer BW, Ersen A, Uribe H, Fadul CE, et al. : Anti-VEGF therapy in pituitary carcinoma. Pituitary 15:445–449, 2012 C4
55. Perrin G, Stevenaert A, Jouanneau E : [Technical aspects and surgical strategy for removal of corticotroph pituitary adenoma]. Neurochirurgie 48:186–214, 2002 C4
56. Petersenn S, Farrall AJ, Block C, Melmed S, Schopohl J, Caron P, et al. : Long-term efficacy and safety of subcutaneous pasireotide in acromegaly : results from an open-ended, multicenter, Phase II extension study. Pituitary:2013 B2
57. Pita-Gutierrez F, Pertega-Diaz S, Pita-Fernandez S, Pena L, Lugo G, Sangiao-Alvarellos S, et al. : Place of preoperative treatment of acromegaly with somatostatin analog on surgical outcome : a systematic review and meta-analysis. PLoS ONE 8:e61523, 2013 C4
58. Potorac I, Petrossians P, Daly AF, Schillo F, Ben Slama C, Nagi S, et al. : Pituitary MRI characteristics in 297 acromegaly patients based on T2-weighted sequences. Endocr Relat Cancer:2015 B2
59. Primeau V, Raftopoulos C, Maiter D : Outcomes of transsphenoidal surgery in prolactinomas : improvement of hormonal control in dopamine agonist-resistant patients. Eur J Endocrinol 166:779–786, 2012 C4
60. Rajasekaran S, Vanderpump M, Baldeweg S, Drake W, Reddy N, Lanyon M, et al. : UK guidelines for the management of pituitary apoplexy. Clin Endocrinol (Oxf) 74:9–20, 2011 A1
61. Randeva HS, Schoebel J, Byrne J, Esiri M, Adams CB, Wass JA : Classical pituitary apoplexy : clinical features, management and outcome. Clin Endocrinol (Oxf) 51:181–188, 1999 C4
62. Raverot G, Vasiljevic A, Raverot V, Lapras V, Favrel V, Trouillas J, et al. : Les adénomes hypophysaires gonadotropes, in EMC. 2014 C4
63. Raverot G, Wierinckx A, Jouanneau E, Auger C, Borson-Chazot F, Lachuer J, et al. : Clinical, hormonal and molecular characterization of pituitary ACTH adenomas without (silent corticotroph adenomas) and with Cushing’s disease. 35–43, 2010 C4
64. Regis J, Castinetti F : Radiosurgery : a useful first-line treatment of prolactinomas ? World Neurosurg 74:103–104, 2010 C4
65. Sasaki R, Murakami M, Okamoto Y, Kono K, Yoden E, Nakajima T, et al. : The efficacy of conventional radiation therapy in the management of pituitary adenoma. Int J Radiat Oncol Biol Phys 47:1337–1345, 2000 C4
66. Sheehan JP, Starke RM, Mathieu D, Young B, Sneed PK, Chiang VL, et al. : Gamma Knife radiosurgery for the management of nonfunctioning pituitary adenomas : a multicenter study. J Neurosurg 119:446–456, 2013 B2
67. Sibal L, Ball SG, Connolly V, James RA, Kane P, Kelly WF, et al. : Pituitary apoplexy : a review of clinical presentation, management and outcome in 45 cases. Pituitary 7:157–163, 2004 C4
68. Storr HL, Alexandraki KI, Martin L, Isidori AM, Kaltsas GA, Monson JP, et al. : Comparisons in the epidemiology, diagnostic features and cure rate by transsphenoidal surgery between paediatric and adult-onset Cushing’s disease. Eur J Endocrinol 164:667–674, 2011 C4
69. Sun DQ, Cheng JJ, Frazier JL, Batra S, Wand G, Kleinberg LR, et al. : Treatment of pituitary adenomas using radiosurgery and radiotherapy : a single center experience and review of literature. Neurosurg Rev 34:181–189, 2010 C4
70. Suzuki C, Maeda M, Hori K, Kozuka Y, Sakuma H, Taki W, et al. : Apparent diffusion coefficient of pituitary macroadenoma evaluated with line-scan diffusion-weighted imaging. J Neuroradiol 34:228–235, 2007 C4
71. Tamasauskas A, Sinkunas K, Bunevicius A, Radziunas A, Skiriute D, Deltuva VP : Transsphenoidal surgery for microprolactinomas in women : results and prognosis. Acta Neurochir (Wien) 154:1889–1893, 2012 C4
72. Tanaka S, Link MJ, Brown PD, Stafford SL, Young WF, Pollock BE : Gamma knife radiosurgery for patients with prolactin-secreting pituitary adenomas. World Neurosurg 74:147–152, 2010 C4
73. Toni R : Ancient views on the hypothalamic-pituitary-thyroid axis : an historical and epistemological perspective. Pituitary 3:83–95, 2000 C4
74. Trouillas J, Delgrange E, Jouanneau E, Maiter D, Guigard MP, Donckier J, et al. : [Prolactinoma in man : clinical and histological characteristics]. Ann Endocrinol (Paris) 61:253–257, 2000 C4
75. Trouillas J, Roy P, Sturm N, Dantony E, Cortet-Rudelli C, Viennet G, et al. : A new prognostic clinicopathological classification of pituitary adenomas : a multicentric case-control study of 410 patients with 8 years post-operative follow-up. Acta Neuropathol 126:123–135, 2013 B2
76. Van Varsseveld NC, Bisschop PH, Biermasz NR, Pereira AM, Fliers E, Drent ML : A long-term follow-up study of eighteen patients with thyrotropin-secreting pituitary adenomas. Clin Endocrinol (Oxf):2013 C4
77. Vlaeminck-Guillem V, Wémeau JL : [Thyroid hormone resistance syndromes : clinical aspects]. Rev Med Interne 20:1114–1122, 1999 C4
78. Vroonen L, Jaffrain-Rea M-L, Petrossians P, Tamagno G, Chanson P, Vilar L, et al. : Prolactinomas resistant to standard doses of cabergoline : a multicenter study of 92 patients. Eur J Endocrinol 167:651–662, 2012 C4
79. Wind JJ, Lonser RR, Nieman LK, DeVroom HL, Chang R, Oldfield EH : The lateralization accuracy of inferior petrosal sinus sampling in 501 patients with Cushing’s disease. J Clin Endocrinol Metab 98:2285–2293, 2013 B2
80. Zada G, Woodmansee WW, Ramkissoon S, Amadio J, Nose V, Laws ER Jr : Atypical pituitary adenomas : incidence, clinical characteristics, and implications. J Neurosurg 114:336–344, 2011 C4
[1] 24
[2] 9,11,49
[3] 25,74
[4] 14,48
[5] 6
[6] 8,76
[7] 62
[8] 11,45
[9] 49
[10] 1,77
[11] 3,61,67
Portfolio
Messages
1. Adénomes hypophysaires , 13 mai 2023, 22:40, par chrisrabeau
Bonjour, homme de 51 ans qui vient de passer en mai 2023 à la chirurgie d’extraction d’un macro adenome ante hypophysaire homogène de 18mm en France, à Tours.
A ce stade et à la lecture de cet excellent document, ma pathologie rentre dans la catégorie des adenomes somatotropes. Des valeurs élevées d Igf1, sans être exagérées, une certaine intolérance aux glucides ainsi que d’autres facteurs (fatigue et douleurs articulaires anormales, maux de tête depuis la majorité...) concordent bien.
Je souffre aussi d’apnées modérées à fortes du sommeil, nécessitant un appareillage depuis 2 ans, un hypoxynemie flagrante de 93 à 83 SPO2 réguliers. Enfin, ingénieur informatique, je souffre depuis quelques années de serieuses dégradations cognitives et memorielles, mal compensées par de l’orthophonie pourtant pratiquées avec sérieux, me rendant au mieux humouristique auprès du public et au pire Roi des phrases absurdes, évidemment sans le vouloir. Il est d’ailleurs assez difficile pour moi d’écrire ceci en ce moment. Je pense que mon très modeste point de vue interieur de cette pathologie pourrait apporter des éléments de solidifications des concepts énoncés ici. Je m’interroge d’ailleurs de ne pas avoir trouvé sur la largeur du Web une quelconque Association des victimes de cette maladie qui semble croître. Ce sera sûrement l’occasion de la fonder.
Je tenais fermement à vous remercier de la remarquable complètude de ce texte datant pourtant de 2015 et que je trouve très à jour.
Je suis totalement ouvert à tous échanges avec ceux qui verront un intérêt potentiel de faire avancer la recherche. Bien à vous.
2. Vraisemblablement micro-adenome hypophissaire 6mm, 27 mai 2023, 04:00, par salma
Je m inquiète je suis âgé de 63ans homme et je vien d à voir ce bilan pouvez vous m expliquer s il ya un traitement et les risques merci