#12. Hémangioblastomes de la fosse postérieure.
Julien ENGELHARDT, M.D.,Hugues LOISEAU, M.D., Ph.D., CHU Bordeaux, mars 2022.
QCM :
1. Concernant les hémangioblastomes, quelles sont les réponses exactes ?
A. Ces tumeurs se développent préférentiellement dans la moelle épinière
B. Les hémangioblastomes du cervelet sont en général liés à la maladie de Von Hippel Lindau
C. Il s’agit de tumeurs survenant préférentiellement chez l’enfant et le jeune adulte
D. L’OMS définit 3 grades histologiques
E. Ces tumeurs sont plus fréquentes chez la femme en âge de procréer
2. Concernant la maladie de Von Hippel-Lindau, quelles sont les réponses exactes ?
A. Il s’agit d’une phacomatose
B. Le gène muté est un proto-oncogène
C. Les hémangioblastomes survenant au cours de cette maladie ont tendance à se développer à un plus jeune âge que dans les formes sporadiques
D. C’est une maladie à transmission autosomique récessive
E. Cette maladie entraîne un risque accru de tumeurs neuro-endocrines pancréatiques
3. Un jeune homme de 36 ans, aux antécédents familiaux de maladie de Von Hippel Lindau, vient vous voir en consultation muni de son IRM. Il s’agit de son premier examen. Son imagerie montre 4 petits hémangioblastomes non kystiques millimétriques. L’examen neurologique est normal.
A. Un diagnostic génétique moléculaire est indispensable pour affirmer qu’il est lui-même atteint de la maladie de Von Hippel Lindau
B. Le diagnostic de maladie de VHL est certain même en l’absence d’autre tumeurs associées
C. Vous lui annoncez qu’il y a un très fort risque (> 2/3) que chacune de ces lésions croissent avec le temps
D. Vous lui proposez une exérèse chirurgicale des lésions accessibles, vu son jeune âge
E. Vous discutez de son dossier en RCP avec les radiothérapeutes, en vue d’une radiothérapie stéréotaxique de ces petites lésions non kystiques, avant qu’elles n’évoluent.
4. Le bilan que vous devez faire réaliser à ce jeune patient comprend :
A. Un examen ophtalmologique avec lampe à fente
B. Un dosage des métanéphrines
C. Un audiogramme
D. Une IRM médullaire
E. Un scanner thoraco-abdomino-pelvien
5. Trois années se sont écoulées. Un jour, votre patient se retrouve hospitalisé dans le service en urgence pour céphalées intenses, vomissements et torticolis. L’imagerie met en évidence une progression d’un des nodules déjà présents, mais surtout l’apparition d’un volumineux kyste péritumoral, compressif.
A. Si votre patient avait été compliant, vous auriez recontrôlé son imagerie tous les ans
B. Vous demandez à vos collègues neuroradiologues une embolisation de la tumeur avant de l’opérer
C. En vue de diminuer le risque de récidive, vous discutez d’une radiothérapie adjuvante post-opératoire, d’autant plus que vous n’avez pas pu peler les parois du kyste
D. Si votre patient était venu aux rendez-vous de suivi, et que vous aviez objectivé à l’imagerie une croissance nodulaire d’un des hémangioblastomes, vous auriez proposé une mise en traitement.
E. Si votre patient était venu aux rendez-vous de suivi, et que vous aviez objectivé à l’imagerie une croissance kystique d’un des hémangioblastomes, vous auriez proposé une mise en traitement.
Introduction
Les hémangioblastomes sont des tumeurs du SNC rares, bénignes (WHO grade 1), essentiellement de l’adulte, pouvant apparaître de manière sporadique (70%) ou associés à la maladie de Von Hippel Lindau (VHL, 30%) (1). Ils comptent pour 2% des tumeurs intra-crâniennes et 2-10% des tumeurs médullaires (1). Leurs localisations préférentielles sont le cervelet puis la moelle épinière et beaucoup moins fréquemment l’étage sus-tentoriel et le tronc cérébral (1).
I. Epidémiologie et histoire naturelle
Le SEER program (Surveillance Epidemiology and End Results), qui couvre 28% de la population des États-Unis, permet d’estimer le taux d’incidence à 0,141 nouveaux cas / 100 000 habitants / an. L’incidence est la plus élevée chez les 60-79 ans (0,246 / 100 000 / an) et la moins élevée chez les enfants (0-19 ans : 0,036 / 100 000 / an). L’âge moyen au diagnostic est de 47 +/- 17 ans. Malgré cette rareté, il s’agit de la tumeur primitive cérébelleuse la plus fréquente de l’adulte. A noter un taux d’incidence légèrement mais significativement plus élevé chez les hommes que les femmes (IRR = 1,2). Le taux de survie globale à 1, 5 et 10 ans est de 95%, 92% et 88,8% respectivement, à nuancer par le caractère unique ou multiple de ces tumeurs (le taux de survie à 5 ans passant de 90 à 85% pour les tumeurs multiples) (1).
L’histoire naturelle des hémangioblastomes dans le cadre de la maladie de VHL est assez bien connue (2). Le sexe masculin est un facteur de risque de charge tumorale augmentée ainsi que de progression tumorale. Cela est cohérant avec le fait que la grossesse n’augmente pas le risque de progression des hémangioblastomes. 72% des patients vont développer de nouveaux hémangioblastomes sur une durée moyenne de 7 ans de suivi. Le risque est d’autant plus élevé en cas de jeune âge, de charge tumorale élevée pré-existante et de délétions (contrairement aux mutations non-sens). La croissance de chaque tumeur est variable : 51% restent stables, 49% grossissent selon trois schémas différents (saltatoire, alternant des périodes de croissance et de quiescence pendant des années : 72%, exponentiel : 22%, linéaire : 6%). Le caractère symptomatique, kystique et le sexe masculin sont les trois facteurs de risque de progression tumorale. Une taille supérieure à 5,6 ; 4,5 et 4 mm respectivement dans le cervelet, la moelle épinière et le tronc cérébral sont associées à un risque de devenir symptomatique avec une spécificité de plus de 90%. La progression tumorale est associée à une mortalité de 25% et pourrait être la cause de décès principale des patients atteint de VHL devant les carcinomes rénaux.
II. Maladie de Von-Hippel-Lindau, bilan
Il s’agit d’une maladie néoplasique systémique caractérisée par un risque accru de développer des hémangioblastomes de l’ensemble du SNC (rétine comprise), des carcinomes à cellules claires du rein (CRCC), des phéochromocytomes et des tumeurs neuroendocrines ou des kystes pancréatiques (TNEP).
Son incidence est estimée à 1 nouveau cas pour 31 000 à 36 000 naissances (3). Le mode de transmission est autosomique dominant, avec une pénétrance de 95% à l’âge de 60 ans (4). Le gène VHL est un gène suppresseur de tumeur porté par le chromosome 3p25. La plupart des patients n’héritent que d’un allèle inactif du gène (3). Il existe une association entre le génotype (type de mutation) et le phénotype permettant de catégoriser les patients atteints de cette maladie en 2 grands groupes (5) : le type I comprend les manifestations du VHL sauf le phéochromocytome, et le type II est divisé en 3 sous-groupes : IIA : risque faible de CRCC et de TNEP, IIB : haut risque de CRCC et de TNEP, IIC : phéochromocytome quasi isolé (pas d’hémangioblastomes ni CRCC).
Les critères diagnostiques de la maladie de VHL sont les suivants (6) :
- En cas d’histoire familiale avérée (sur plusieurs générations), la présence d’une seule lésion associée au VHL suffit à poser le diagnostic (80% des cas)
- En l’absence d’antécédents familiaux (20%), la présence d’un hémangioblastome du SNC (dont la rétine) associée à une autre lésion du spectre du VHL pose le diagnostic.
- En cas de doute, un test génétique est indiqué. A noter que dans les 20% de cas de VHL de novo (sans histoire familiale connue), la confirmation génétique peut être mise en défaut du fait d’un mosaïcisme expliquant que l’ensemble des leucocytes ne présente pas la mutation.
Le bilan initial comprend un fond d’œil dilaté, un audiogramme, une IRM de l’encéphale et de la moelle spinale, une IRM abdominale, un dosage des catécholamines (métanéphrines) plasmatiques et/ou urinaires et une demande de consultation génétique.
III. Anatomopathologie et profil génétique (7)
Ces tumeurs sont classées comme des tumeurs mésenchymateuses non méningothéliales.
Ces tumeurs restent histologiquement bénignes, de grade I de l’OMS. Il existe une double prolifération, vasculaire (capillaires de structure normale), et cellulaire inter-vasculaire fait de travées de cellules arrondies ou polygonales de grande taille avec un noyau central sphérique et un cytoplasme clair riche en glycogène et contenant de petites gouttelettes lipidiques ("stromal cells")
Macroscopiquement, il s’agit de tumeurs richement vascularisées, partiellement kystiques (60%) ou entièrement solides (40%). L’apparence classique est celle d’un kyste rempli d’un liquide jaune clair (protéique) avec un nodule mural rouge vif proche de la surface piale. Les nodules peuvent être millimétriques.
Le gène suppresseur de tumeur VHL est inactivé à la fois dans les hémangioglastomes associés à la maladie éponyme, mais également dans la plupart (78%) des formes sporadiques. Dans les hémangioblastomes familiaux (VHL), l’inactivation bi-allélique est la règle, contrairement aux tumeurs sporadiques. Aucun autre gène n’a pour l’instant été mis en cause dans le développement de ces tumeurs. La perte de fonction de VHL entraîne une « upregulation » des gènes sensibles à l’hypoxie, tels que ceux codant pour le VEGF et l’EPO, en l’absence d’hypoxie tumorale (condition appelée pseudo-hypoxie). Cela explique le caractère très richement vascularisé de ces tumeurs, ainsi que la polyglobulie par sécrétion inappropriée d’EPO retrouvée chez certains malades.
IV. Clinique
Les signes cliniques d’un hémangioblastome cérébelleux sont ceux de n’importe quel syndrome de masse de la fosse postérieure. Ces signes peuvent être bruyants et rapidement progressifs avec un risque de décompensation rapide en cas de kyste volumineux. Rarement, le mode de révélation peut être aigu, lié à un hématome intra-cérébelleux par hémorragie intra-tumorale.
V. Paraclinique
Le diagnostic est en général aisé chez les patients connus et suivis pour une maladie de VHL. Chez les patients non connus ou non atteints de cette maladie, la découverte d’un hémangioblastome doit faire réaliser un bilan à la recherche d’autres lésions du spectre de la maladie VHL (cf partie II. )
Le bilan d’imagerie comprend :
- Un scanner cérébral dans les modes de révélations aigus (urgences) : lésion isodense prenant intensément le contraste, parfois kystique
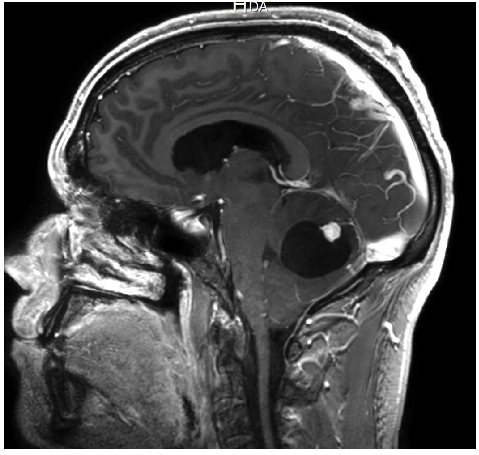
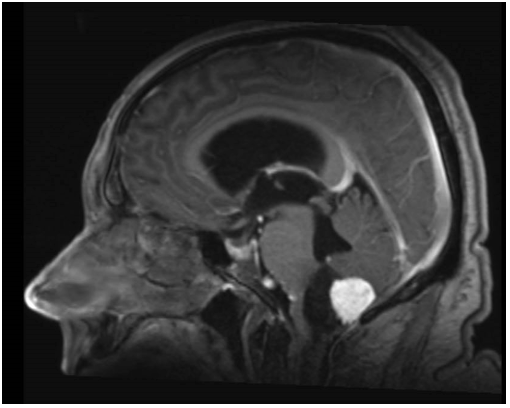
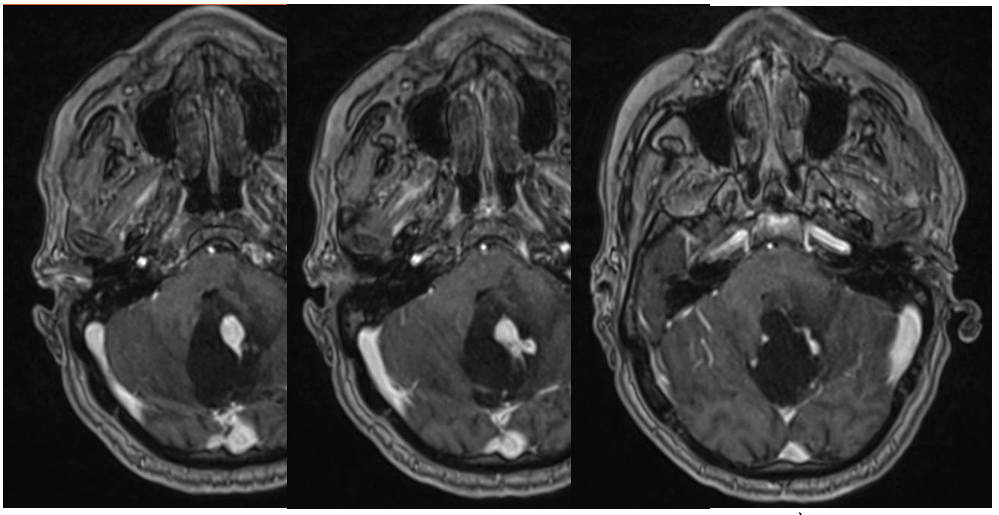
- Une IRM encéphalique (figures 1 à 3) : examen de référence. Lésion iso-intense en T1, prenant le contraste intensément. Pas de prise de contraste des parois du kyste (diagnostic différentiel avec astrocytome pilocytique). Rechercher des images vasculaires (flow void en T2) en périphérie de la lésion ainsi que des dépôts d’hémosidérine (T2*) témoins de micro-saignements anciens.
- Rarement, une angiographie vertébrale en pré-opératoire peut être utile dans les formes « purement » kystiques sans nodule mural visualisé à l’IRM.
A noter 4 patterns différents : classiquement, un kyste avasculaire avec un nodule mural unique (figure 1), mais aussi : des nodules multiples, séparés les uns des autres sur les parois du kyste (figure 3) ; la forme nodulaire (« hémangioblastome plein », figure 2) ; et exceptionnellement un kyste intra-tumoral (toute la paroi du kyste est vasculaire : paroi en général plus épaisse et prenant le contraste, diagnostic différentiel avec un astrocytome) (8).
En dehors du bilan d’imagerie, l’hémogramme recherche une polyglobulie (très rare mais classiquement cité dans les publications) et dans les cas suspects (à l’interrogatoire, examen ou antécédents familiaux), un dosage des catécholamines est indiqué pour dépister un phéochromocytome.
VI. Principes de prise en charge
Indications de traitement
La chirurgie reste le traitement de référence. De manière générale, seule les lésions symptomatiques, ou menaçantes sur le plan radiologique, constituent une indication thérapeutique, d’autant plus en cas de maladie de VHL. Dans ce cas précis, le but est de diminuer au maximum le nombre de procédures chirurgicales dans la vie du patient car les tumeurs associées à la maladie de VHL sont multiples et récidivantes, avec une histoire naturelle « biphasique » alternant phases de croissance et de quiescence. Les lésions difficilement accessibles et pauci symptomatiques peuvent être surveillées en première intention. Une attention particulière doit être portée aux hémangioblastomes solides supra-centimétrique de la fosse postérieure, qui devraient être réséqués en première intention le plus tôt possible dans leur évolution, du fait de leur potentiel de croissance rapide et des difficultés (proportionnelles à leur taille) rencontrées dans l’exérèse chirurgicale.
Traitement chirurgical : cf infra
Radiothérapie fractionnée (9)
Malgré des résultats montrant un contrôle tumoral dose-dépendant, le taux de contrôle moyen dans la littérature est estimé entre 27 et 57%. En contrepartie, un certain nombre d’effets indésirables aigus et à long terme ont été rapportés. Enfin, il a été montré que la radiothérapie fractionnée ne permettait pas de prévenir la récidive en cas d’exérèse incomplète.
Radiothérapie stéréotaxique (SRS) (2,3,9,10)
La SRS est connue pour assurer un meilleur contrôle que la radiothérapie conventionnelle pour les tumeurs bénignes et très vascularisées, ce qui théoriquement fait des hémangioblastomes une indication de choix. Dans la plus grande étude internationale portant sur 189 patients et 517 tumeurs (rétrospective), le taux de contrôle tumoral était de 92% à 3 ans, 89% à 5 ans, 79% à 10 ans, avec un taux de survie globale de 94% à 3 ans, 90% à 5 ans et 74 à 10 ans. Une méta-analyse récente, incluant 14 études, a montré que la PFS à 5 ans était de 88,4%. Cependant, les résultats à plus long terme ne sont pas connus, et la PFS à 10 ans varie entre 50 et 96% selon les séries. En revanche, le caractère kystique des hémangioblastomes est un frein à l’utilisation de la SRS et en diminue l’efficacité. En effet, dans une série 32 patients et 74 lésions, dont 8 patients avec des lésions kystiques, la PFS pour les tumeurs solides à 1, 3 et 5 ans étant de 98%, 98% et 94,9% comparé à 88,9%, 76,2% et 61% pour les tumeurs kystiques. D’autres séries ont rapporté une tendance des kystes à progresser après SRS, indiquant une chirurgie de sauvetage.
Les complications de la SRS sont en général retardées, avec une morbidité médiane de 3,1% sur 23 études. Les complications principales sont la radionécrose et la formation ou majoration d’un œdème périlésionnel entraînant céphalées, nausées et vomissements voire une hydrocéphalie aiguë obstructive.
Au total, les études publiées étant de faible niveau de preuve, sans groupes contrôles ni randomisation, les effets présumés de la SRS peuvent aussi être attribués à une phase de quiescence concomitante de la croissance tumorale. Ainsi, en l’état actuel des connaissances, la SRS n’est indiquée qu’en 2ème intention, dans le cas d’un hémangioblastome requérant un traitement et non accessible chirurgicalement. Il n’y pas d’indication à traiter de manière prophylactique des hémangioblastomes multiples asymptomatiques.
Chimiothérapies (11,12)
A l’heure actuelle, aucun traitement médical n’est validé. Plusieurs molécules ont été testées (inhibiteurs des récepteurs à tyrosine kinase : semaxanib, sunitinib, vatalanib en partie efficaces sur certains cancers du rein mais décevants sur les hémangioblastomes ; immuno-modulateurs tels que la thalidomide et l’interféron alpha 2-a, anti-VEGF tel que le bevacizumab (Avastin ©) en injections intra-vitréennes pour les hémangioblastomes rétiniens bien que le niveau de preuve reste faible). Actuellement, l’attention se porte sur les effets des bêtabloquants et notamment du propranolol via son action bloquant les récepteurs bêta 2 adrénergiques. Après avoir montré une certaine efficacité sur les hémangioblastomes rétiniens, l’EMA (agence européenne du médicament) a inscrit le propranolol en 2017 comme médicament orphelin pour le traitement des tumeurs du spectre de la maladie de VHL. Cependant, l’utilisation reste limitée par les effets indésirables cardio-vasculaires chez des sujets normotendus dus à l’effet anti bêta-1, d’où le développement en cours d’un antagoniste spécifique bêta-2 (ICI 118,551).
VII. Traitement chirurgical
Ressource à consulter :
https://www.neurosurgicalatlas.com/volumes/brain-tumors/supratentorial-and-posterior-fossa-tumors/hemangioblastoma?highlight=hema
Planning
L’imagerie pré-opératoire doit être soigneusement analysée : relation avec le plancher du IVème ventricule et avec la PICA qui donne dans la majorité des cas les afférences principales à la tumeur. Dans les cas de maladie de VHL, il est important de garder à l’esprit que le patient sera réopéré au cours de sa vie. De l’incision jusqu’à la fermeture, tout doit être planifié en ce sens. On préfèrera ainsi une incision médiane permettant de ré-aborder la fosse postérieure ultérieurement. De plus, en cas d’hémangioblastome cervical ou bulbaire aisément accessible (même asymptomatique) associé à un hémangioblastome cérébelleux, l’exérèse pourra être discutée et considérée dans le même temps, toujours dans le but de limiter le nombre de ré-intervention.
Procédures préparatoires à l’exérèse
En cas d’hydrocéphalie aiguë menaçante, une ventriculocisternostomie peut être réalisée dans l’attente de l’exérèse de la tumeur.
En général, l’embolisation pré-opératoire n’est pas indiquée, sauf en cas de certaines lésions volumineuses et sans kyste. L’embolisation permettrait de diminuer l’apport vasculaire et rendre la chirurgie plus aisée notamment en cas d’apport artériel antérieur pouvant être responsables d’un saignement important avant d’avoir pu les contrôler. L’embolisation pré-opératoire des hémangioblastomes médullaires peut également se discuter.
Procédure chirurgicale
Les hémangioblastomes doivent être abordés et réséquées selon les mêmes principes que la chirurgie des MAV (malformations artério-veineuses). Ainsi, leur exposition doit être généreuse et doit permettre d’individualiser et contrôler les afférences artérielles et les veines de drainage. L’existence d’un kyste facilite la chirurgie grandement. Une ouverture première de kyste permet d’obtenir une détente cérébelleuse et commencer à retrouver le plan de clivage. L’anatomie vasculaire peut être encore mieux individualisée grâce à une vidéo-angiographie au vert d’indocyanine (ICG)13. L’ICG peut également être très utile en cas de chirurgie pour une récidive, où des nodules muraux millimétriques peuvent être moins facilement visibles dans la gliose. Comme pour les MAV, les afférences doivent être déconnectées, et le nodule ne doit pas être abordé avant sa dévascularisation. Quelque soit le volume de la léion, sa réduction par débulking n’est pas envisageable, et le « dégonflement » de la tumeur par le contrôle des afférences artérielles est la seule option. En cas d’entrée impromptue dans le nodule, l’hémostase par tamponnement plus ou moins application d’hémostatiques tel que le Floseal © reste la meilleure option. Une fois que les afférences sont coagulées, la ou les veines de drainages sont sacrifiées, et le nodule est réséqué en bloc en entrant dans le plan gliotique entre la substance blanche et la tumeur. Contrairement aux MAV, il n’existe en général pas de petites afférences profondes provenant de la substance blanche, rendant l’exérèse plus aisée. Sauf cas particulier (cf supra), les parois du kyste ne sont pas touchées, et simplement inspectées pour s’assurer de l’exérèse complète et de l’absence de petits nodules satellites.
Hémangioblastomes solides
Ces tumeurs font partie, chirurgicalement, de la catégorie des MAV. Il s’agit d’un amas de capillaires facilement hémorragiques, et dont l’hémostase ne peut pas être obtenue par coagulation bipolaire. Quelque soit le volume de la masse, sa réduction par débulking n’est pas envisageable, et le « dégonflement » de la tumeur par le contrôle des afférences artérielles est la seule option. Cependant, les afférences sont rarement toutes accessibles à la phase initiale de la chirurgie, d’où l’intérêt primordial de l’embolisation pré-opératoire. En cas d’hémangioblastomes géants, une approche en plusieurs étapes peut être nécessaire pour éviter un gonflement cérébelleux majeur lié au sacrifice dans le même temps de l’ensemble de la vascularisation. Une autre difficulté réside dans le fait que le chirurgien fait face en premier aux veines de drainages, souvent accolées à la tumeur, et qui doivent être absolument respectées jusqu’à la dévascularisation complète afin d’éviter une hémorragie tumorale difficilement contrôlable.
Hémostase
Un hématome de fosse postérieure menaçant le pronostic vital peut se développer rapidement du fait d’une hypertension post-opératoire incontrôlée, du fait du caractère fortement vasculaire de ces tumeurs. L’hémostase doit être méticuleusement vérifiée en fin d’intervention.
VIII. Suivi thérapeutique
Le suivi des patients atteints de la maladie de VHL est sujet à variations selon les pays. En France, le Réseau National de Référence pour les Cancers rares de l’adulte (PREDIR) et l’association VHL France ont publié des recommandations : https://www.vhlfrance.org/recommandations-de-suivi-clinique-vhl/.
En cas d’hémangioblastome sporadique, il n’existe pas de recommandation particulière. En pratique, une IRM peut être réalisée à 3 mois post-opératoire, puis à 1 an, 3 ans et 5 ans. Le taux de récidive en cas d’exérèse complète est pratiquement nul à 5 ans3. Il est important de noter que certains groupes préconisent un dépistage génétique chez tous les patients diagnostiqués avec un hémangioblastome sans maladie de VHL connue (3).
Points forts :
- Tumeurs bénignes solides ou kystiques du SNC et de la rétine, richement vascularisées et non infiltrantes
- La plus fréquente des tumeurs primitives du SNC de l’adulte dans la fosse postérieure
- Peut survenir dans le cadre d’une maladie de Von Hippel Lindau
- Peut être associée à une polyglobulie
- La maladie de Von Hippel Lindau est une maladie à transmission autosomique dominante, par perte d’expression du gène suppresseur de tumeur VHL
- L’âge de début est variable, mais la pénétrance est de 95% à 60 ans
- L’âge moyen de développement des hémangioblastomes associés au VHL est avancé de 10 ans par rapport aux tumeurs sporadiques
- Compte-tenu de l’histoire naturelle difficilement prédictible (1 tumeur /2 ne grossirait pas, croissance biphasique ou saltatoire), seules les lésion symptomatiques ou menaçantes sur le plan radiologique doivent être traitées.
Réponses aux QCM
QCM 1. Concernant les hémangioblastomes, quelles sont les réponses exactes ?
A. Ces tumeurs se développent préférentiellement dans la moelle épinière
B. Les hémangioblastomes du cervelet sont en général liés à la maladie de Von Hippel Lindau
C. Il s’agit de tumeurs survenant préférentiellement chez l’enfant et le jeune adulte
D. L’OMS définit 3 grades histologiques
E. Ces tumeurs sont plus fréquentes chez la femme en âge de procréer
QCM 2. Concernant la maladie de Von Hippel-Lindau, quelles sont les réponses exactes ?
A. Il s’agit d’une phacomatose
B. Le gène muté est un proto-oncogène
C. Les hémangioblastomes survenant au cours de cette maladie ont tendance à se développer à un plus jeune âge que dans les formes sporadiques
D. C’est une maladie à transmission autosomique récessive
E. Cette maladie entraîne un risque accru de tumeurs neuro-endocrines pancréatiques
QCM 3. Un jeune homme de 36 ans, aux antécédents familiaux de maladie de Von Hippel Lindau, vient vous voir en consultation muni de son IRM. Il s’agit de son premier examen. Son imagerie montre 4 petits hémangioblastomes non kystiques millimétriques. L’examen neurologique est normal.
A. Un diagnostic génétique moléculaire est indispensable pour affirmer qu’il est lui-même atteint de la maladie de Von Hippel Lindau
B. Le diagnostic de maladie de VHL est certain même en l’absence d’autre tumeurs associées
C. Vous lui annoncez qu’il y a un très fort risque (> 2/3) que chacune de ces lésions croissent avec le temps
D. Vous lui proposez une exérèse chirurgicale des lésions accessibles, vu son jeune âge
E. Vous discutez de son dossier en RCP avec les radiothérapeutes, en vue d’une radiothérapie stéréotaxique de ces petites lésions non kystiques, avant qu’elles n’évoluent.
QCM 4. Le bilan que vous devez faire réaliser à ce jeune patient comprend :
A. Un examen ophtalmologique avec lampe à fente
B. Un dosage des métanéphrines
C. Un audiogramme
D. Une IRM médullaire
E. Un scanner thoraco-abdomino-pelvien
QCM 5. Trois années se sont écoulées. Un jour, votre patient se retrouve hospitalisé dans le service en urgence pour céphalées intenses, vomissements et torticolis. L’imagerie met en évidence une progression d’un des nodules déjà présents (restant centimétrique), mais surtout l’apparition d’un volumineux kyste péritumoral, compressif.
A. Si votre patient avait été compliant, vous auriez recontrôlé son imagerie tous les ans
B. Vous demandez à vos collègues neuroradiologues une embolisation de la tumeur avant de l’opérer
C. En vue de diminuer le risque de récidive, vous discutez d’une radiothérapie adjuvante post-opératoire, d’autant plus que vous n’avez pas pu peler les parois du kyste
D. Si votre patient était venu aux rendez-vous de suivi, et que vous aviez objectivé à l’imagerie une croissance nodulaire d’un des hémangioblastomes, vous auriez proposé une mise en traitement.
E. Si votre patient était venu aux rendez-vous de suivi, et que vous aviez objectivé à l’imagerie une croissance kystique d’un des hémangioblastomes, vous auriez proposé une mise en traitement.
Références annotées
- Louis, D. N. et al. The 2016 World Health Organization classification of tumors of the central nervous system : A summary. Acta Neuropathol 131, 803–820 (2016).
Description histologique et physiopathologie moléculaire et génétique. - Klingler, J.-H. et al. Hemangioblastoma and von Hippel-Lindau disease : genetic background, spectrum of disease, and neurosurgical treatment. Childs Nerv Syst 36, 2537–2552 (2020).
Article de synthèse très complet sur la maladie de Von Hippel Lindau - Lonser, R. R. et al. Prospective natural history study of central nervous system hemangioblastomas in von Hippel-Lindau disease. J Neurosurg 120, 1055–1062 (2014).
Etude prospective ayant permis de décrire précisément l’évolution des hémangioblastomes dans le cadre de la maladie de VHL.
Références :
1. Yin, X. et al. Incidence, Prognostic Factors and Survival for Hemangioblastoma of the Central Nervous System : Analysis Based on the Surveillance, Epidemiology, and End Results Database. Front Oncol 10, 570103 (2020).
2. Lonser, R. R. et al. Prospective natural history study of central nervous system hemangioblastomas in von Hippel-Lindau disease. J Neurosurg 120, 1055–1062 (2014).
3. Klingler, J.-H. et al. Hemangioblastoma and von Hippel-Lindau disease : genetic background, spectrum of disease, and neurosurgical treatment. Childs Nerv Syst 36, 2537–2552 (2020).
4. Catapano, D. et al. Hemangioblastomas of central nervous system : molecular genetic analysis and clinical management. Neurosurgery 56, 1215–1221 ; discussion 1221 (2005).
5. Friedrich, C. A. Genotype-phenotype correlation in von Hippel-Lindau syndrome. Hum Mol Genet 10, 763–767 (2001).
6. Lonser, R. R. et al. von Hippel-Lindau disease. Lancet 361, 2059–2067 (2003).
7. Louis, D. N. et al. The 2016 World Health Organization classification of tumors of the central nervous system : A summary. Acta Neuropathol 131, 803–820 (2016).
8. Fs, B., Jk, L., Ss, C. & Dw, F. Recurrent cerebellar hemangioblastoma with enhancing tumor in the cyst wall : case report. Neurosurgery 62, (2008).
9. Pan, J., Jabarkheel, R., Huang, Y., Ho, A. & Chang, S. D. Stereotactic radiosurgery for central nervous system hemangioblastoma : systematic review and meta-analysis. J Neurooncol 137, 11–22 (2018).
10. Liebenow, B. et al. Gamma Knife Stereotactic Radiosurgery favorably changes the clinical course of hemangioblastoma growth in von Hippel-Lindau and sporadic patients. J Neurooncol 142, 471–478 (2019).
11. Cuesta, A. M. et al. The β2-adrenergic receptor antagonist ICI-118,551 blocks the constitutively activated HIF signalling in hemangioblastomas from von Hippel-Lindau disease. Sci Rep 9, 10062 (2019).
12. Capitanio, J. F., Mazza, E., Motta, M., Mortini, P. & Reni, M. Mechanisms, indications and results of salvage systemic therapy for sporadic and von Hippel-Lindau related hemangioblastomas of the central nervous system. Crit Rev Oncol Hematol 86, 69–84 (2013).
13. Hwang, S. W., Malek, A. M., Schapiro, R. & Wu, J. K. Intraoperative use of indocyanine green fluorescence videography for resection of a spinal cord hemangioblastoma. Neurosurgery 67, ons300-303 ; discussion ons303 (2010).